PHOTODYNAMIC THERAPY and SIGNAL TRANSDUCTION
Anatoly B. Uzdensky
Department of Biophysics and Biocybernetics
Southern Federal University
Rostov-on-Don, 344090, Russia
auzd@yandex.ru
Photodynamic therapy (PDT) is based on the photoinduced generation of highly cytotoxic singlet oxygen, and other reactive oxygen species (ROS), which induce oxidative cell injury that leads to necrotic or apoptotic cell death. Photodynamic killing of pathologically changed cells provides a treatment for cancer and some non-cancerous diseases. Cell death processes are known to be controlled by cell signaling systems. Therefore, the modification of signaling pathways may modulate the cellular response to PDT. The present chapter is devoted to data on mechanisms regulating the cellular response to photodynamic injury. [See Table 1 for a list of the most frequent abbreviations.]
1. The Principal Signaling Pathways
The main finding in modern cell biology is the discovery of a complex cell regulation system consisting of many interacting signaling pathways leading from the cell surface receptors into the cytoplasm, further into the nucleus, and back to the cytoplasm, the cell surface and the extra cellular medium. This system consists of numerous intercellular signaling molecules, surface receptors, cytoplasmic signaling cascades, transcription factors and executioner proteins, which altogether determine a cell's response to external impacts. Some of these factors regulate cell survival, and others control cell death. Numerous, if not all, signaling pathways take part in the cellular responses to PDT.
Everybody who starts to study cell signaling processes meets two significant problems: (i) the huge complexity of the cell signaling system, and (ii) imperfect scientific terminology. The number of cellular proteins is estimated to be about 105. The most of them participate in intracellular signaling and regulation. Only a small fraction of the signaling proteins have been identified so far, and their molecular functions are often not determined. The signaling proteins usually form protein pathways or cascades aimed to execute various cellular functions. However, not all components of these pathways are determined, and their activity is not well defined. The signaling pathways may control different sets of effector proteins in different cell lines or even in the same cells under different conditions. These effector proteins form the physiological state of the cell. But their number and activity are unknown. The interaction of different signaling pathways strongly complicates the issue.
Another problem is the absence of any system for naming the signaling proteins. The latter are mainly senseless abbreviations. Since nobody can keep in mind about 105 such names, researchers working in different fields hardly understand each other. Moreover, some proteins that were discovered several times in different organisms may have different names. But later, when they are identified as the same protein, their names may consist of all primary names, for example, JNK/SAPK, or p21/Cip1/WAF. In the present chapter, I consider only several tens of known signaling proteins that are believed to play central roles in cellular functions. The major known signaling pathways and potential cellular PDT targets are schematically presented at Figure1. Extracellular signaling molecules (hormones, neuromediators, cytokines, or growth factors) are recognized by receptors coupled to G-proteins (RCGP), or by receptor tyrosine kinases (RTK). Extracellular matrix or adjacent cells are recognized by the cell adhesion receptors (integrins, cadherins). There are several signaling pathways in the cell leading from these receptors into the cytoplasm and the nucleus.
The calcium pathway is initiated by Ca2+ influx through the plasma membrane (PM), or by Ca2+ release from Ca2+-storing organelles: endoplasmic reticulum (ER) or mitochondria. The release of Ca2+ from ER may be initiated either by RCGP or by RTK. Ca2+ accumulation in the cytosol activates the downstream Ca2+-dependent signaling proteins, such as calmodulin (CaM), calmodulin-dependent kinase II (CaMKII), protein kinase C (PKC), etc.
In the cAMP signaling pathway, ligand binding to RCGP activates G-protein, which stimulates adenylate cyclase (AC) to produce cyclic adenosine monophosphate (cAMP). cAMP-dependent protein kinase A (PKA) then activates diverse cellular reactions.
Receptor tyrosine kinase may initiate the signaling pathways mediated by: (i) phospholipase C and Ca2+; (ii) phosphatydilinositol 3-kinase (PI3K) and protein kinase B/Akt; (iii) mitogen-activated protein kinases (MAPKs). There are three well-characterized MAP kinases in the MAPK family: extracellularly regulated kinase (ERK), c-Jun terminal kinase (JNK), and protein kinase p38. ERKs are stimulated by cytokines or other chemical signals. They regulate cell proliferation and survival. JNK and p38 are involved in cell reactions to environmental stress: division, survival, or apoptosis.
Integrins, cell adhesion receptors, may activate focal adhesion kinase that regulates cytoskeleton remodeling, cell shape and motility. All these protein kinases regulate numerous effector proteins and transcription factors, which determine cell responses to external impacts. Each cell has an individual set of such proteins that determine its physiological state and provide specific responses.
2. Possible ROS Sensors
Oxidative stress is associated not only with general cell injury, but also with up-regulation of some cellular proteins. These generally include cell protection enzymes (catalase, superoxide dismutase, hemoxygenase-1, ferritin, glutathione peroxidase, glutathione reductase, quinone reductase, thioredoxin, thioredoxin reductase, and cyclooxygenase-2), proteins involved in intercellular interactions, and transcription factors (AP-1, ATF/CREB, ETS, C/EBP, NF-κB) (Turpaev, 2002). PDT activates or up-regulates diverse proteins involved in cell protection (chaperons, superoxide dismutase, heme oxygenase-1), signal transduction and transcription control (cyclooxygenase-2, p38-MAPK, JNK, FAK, PI 3-kinase, Akt/PKB, AP-1, NF-κB), and apoptosis (caspases, Bcl-2 family proteins, nucleases). This is evidently mediated by the cellular regulatory systems (Oleinick et al., 2002; Almeida et al., 2004; Castano et al., 2005; Nowis et al., 2005; Uzdensky, 2008). The primary event in this pathway is recognition of PDT-generated ROS. This suggests the presence of specific ROS sensors in the cell. Alternatively, the cell should recognize results of gross damage such as ionic disbalance, impairment of the energy metabolism, and destruction of cellular constituents.
Relatively long-lived ROS, like hydrogen peroxide (H2O2) and superoxide-anion (O2.-), are produced as minor by-products of oxidative phosphorylation in normal cells. In stress conditions, their production is significantly enlarged (Skulachev, 1999). Short-lived singlet oxygen 1O2, which is produced during PDT, is two orders more active. However, unlike H2O2 and O2.-, animal cells don't meet singlet oxygen in their life, and possibly don't need in specific 1O2 sensors. Nevertheless, the complex cell response to oxidative stress involving diverse signaling pathways implies the presence of ROS sensors reacting to either changes in the cellular redox potential, or peroxide accumulation, or general ROS level. Such sensors have not yet been found, but potential candidates for this role are discussed in the literature.
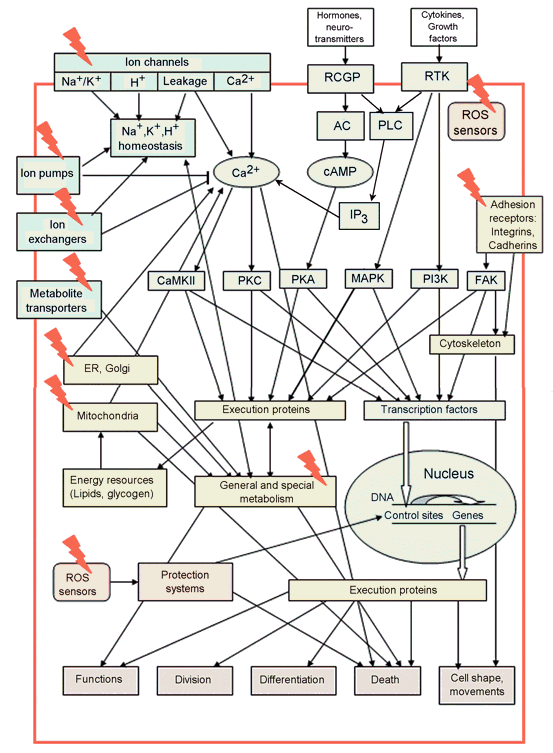
Figure 1. Main cell signaling pathways and cell functions they control. The potential PDT targets are shown by red lightning (Adapted from: Uzdensky, 2008). AC: adenylate cyclase, CaMKII: calmodulin-dependent protein kinase II, ER: endoplasmic reticulum, FAK: focal adhesion kinase, IP3: inositol 1,4,5-trisphosphate, MAPK: mitogen-activated protein kinase, PI3K: phosphatidylinositol 3-kinase, PKA: protein kinase A, PKC: protein kinase C, PLC: phospholipase C, RCGP: receptors coupled to G-proteins, RTK: receptor tyrosine kinase.
Some phospholipid metabolites such as arachidonic acid, diacylglycerol, or ceramide are signaling messengers. The products of 1O2-mediated oxidation of unsaturated lipids such as hydroperoxides (LOOH) and/or radical species (L., LOOH., OLOO., or OLO.) may signal about oxidative cell damage. Long-lived hydroperoxides rather than short-lived radical products can serve as early signaling mediators in photooxidative stress. LOOH transfer between cellular membranes may spread the peroxidative stress throughout the cell. Indirect mechanisms relating lipid peroxides to signaling processes may be also taken into account. For example, phospholipase A2 (PLA2) is more active on peroxidized than normal membranes (Girotti, Kriska, 2004). However, the constitutive antioxidant systems, including lipid antioxidants and ROS detoxication enzymes (catalase, superoxide dismutase, glutathione-dependent peroxidases), prevent or repair oxidative damage in the cell. Only when their antioxidant capacity becomes insufficient for cell protection, the signaling processes may ether induce cell protection mechanisms or cause cell death.
The possible specific ROS sensors:
(i) Some bacteria contain two specific ROS signaling proteins, SoxR and OxyR. In normal cells they await activation. OxyR is activated by low concentrations of H2O2, whereas SoxR is inactivated by O.- and NO.. These ROS induce the formation of intramolecular disulfide bonds that may be reversed by glutaredoxin, which in turn is reversed by glutathione (GSH). SoxR contains two [2Fe-2S] clusters that presumably function as ROS sensors. Activated SoxR stimulates transcription factor SoxS, which triggers the transcription of genes involved in cell defense: catalase, superoxide dismutase, alkyl hydroperoxidase, glutathione reductase. Such elegant ROS sensors are not found in eukaryotes, which probably use more complicated redox-sensitive signaling systems.
(ii) Enzymes with essential thiol groups in the active center are easily inactivated by singlet oxygen. They may link a primary oxidative injury to signal transduction pathways. Tyrosine protein phosphatase (TPP), small GTPases such as Ras, and mitochondrial adenine nucleotide translocator (ANT) are among such proteins. Protein phosphatases contain a highly conserved cystein in the active center. Hensley et al. (2000) suggested that H2O2 inactivates TPP through the formation of the sulfenic acid intermediate TPP-SOH. In the presence of glutathione a mixed glutathione intermediate TPP-S-SG is formed. TPP may be regenerated by glutathione or thioredoxin, which catalyse the reduction of exposed ─S─S─ bridges. Similar reactions may be involved in 1O2-induced TPP inactivation. PDT-induced inhibition of TPP prolongs tyrosine phosphorylation and, therefore, signaling action of phosphorylated enzymes.
ANT plays the significant role in mitochondrial permeability transition and apoptosis. It is more sensitive to photodynamic injury than other mitochondrial proteins. PDT-induced inactivation of ANT is mediated by photooxidation of thiol groups. Its irreversible photodamage occurs before loss of mitochondrial transmembrane potential (ΔΨm), and Ca2+ release from mitochondria (Belzacq et al., 2001).
The cysteine-118 residue in the active center of Ras, a small GTPase that links receptor tyrosine kinases to the MAPK signaling cascade, can be regulated in a redox-dependent mode. This influences nucleotide exchange and Ras-dependent signaling (Finkel, 1998).
(iii) Thioredoxin, a multifunctional redox-sensitive disulfide reductase, may also serve as a sensor of ROS, especially H2O2. Reduced thioredoxin forms an inactive complex with MAP kinase kinase kinase ASK1 (apoptosis signal-regulating kinase-1). After oxidation by H2O2, this complex is disrupted, and free ASK1 initiates MAPK signaling cascades (Fig. 2) including MAP kinase kinases MKK 3, 4, 6 and 7, and downstream MAP kinases p38 and JNK that in turn activate different transcription factors: CREB, ATF-2, c-Jun, etc (Turpaev, 2002). The transcription factor NF-κB is activated by oxidized thioredoxin, whereas reduced thioredoxin inhibits it. Therefore, NF-κB is also involved in the thioredoxin-mediated signaling pathway.
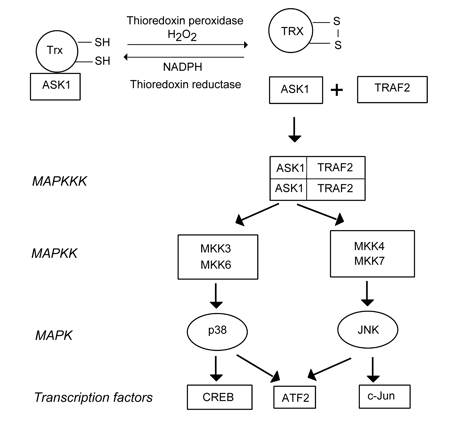
Figure 2. The possible mechanism of ROS sensing by the thioredoxin/ASK1 complex. ASK1: Apoptosis signal-regulating kinase-1, ATF2: activating transcription factor 2, CREB: CRE-binding protein, JNK: c-Jun terminal kinase, MKK: MAP kinase kinase, TRAF2: TNR receptor-associated factor 2, Trx: thioredoxin (From: Turpaev, 2002).(iv) NF-κB may serve as a ROS sensor by itself. PDT-induced 1O2 and other ROS may directly activate NF-κB by phosphorylation of its inhibitory subunit IκB (Matroule, Piette, 2000).
(v) Intracellular oxygen sensors such as hypoxia-inducible transcription factor HIF-1 may also react to PDT-induced rapid oxygen depletion. HIF-1 activation by mROS generated in mitochondria may be a basis of oxygen sensing. Cystein residues involved in redox regulation of HIF-1 are regulated by thioredoxin and REF-1 (redox factor 1). HIF-1 cooperates with the transcription factor AP-1 in the control of cell metabolism and survival.
Thus, there is no single ROS sensor found in cells. Multiple ROS sensors probably function.
3. The Role of Mitochondria in PDT-Induced Apoptosis
Mitochondria are very sensitive to PDT. The primary PDT-induced ROS generation is supplemented by the secondary production of ROS (mROS) in mitochondria with impaired electron transport. The limited generation of superoxide-anion (O2.-) occurs in normal cells due to a one-electron oxygen reduction that occurs mostly at the level of flavins. However, generation of O2.- and other mROS is strongly enhanced when oxidative phosphorylation is uncoupled and coenzyme Q transfers excessive electrons directly to O2 (Skulachev, 1999). The same events occur during PDT.
PDT inhibits diverse bioenergetic enzymes including succinate dehydrogenase, cytochrome c oxidase, H+-ATP-synthase, ANT, and organic acid carriers. As a result of the PDT-induced lipid peroxidation and photodamage of mitochondrial proteins, electron transport is impaired, oxidative phosphorylation is uncoupled, trespiratory control is lost; the ADP/O ratio and mitochondrial transmembrane potential (ΔΨm) is dropped, and ATP synthesis is inhibited. However, photoinjury of mitochondria is not sufficient for complete ATP depletion. A full ATP drop occurs when PDT is combined with the inhibition of glycolysis.
Mitochondria generally play a central role in apoptosis initiation. Cytochrome c (Cyt c) is the main conductor in the apoptotic orchestra. It resides in the mitochondrial intermembrane space (IMS), where it binds to cardiolipin at the outer surface of the inner mitochondrial membrane. Mitochondrial injury causes hyperproduction of ROS inside the mitochondria (mROS), peroxidation of cardiolipin, and mobilization of Cyt c. Then Cyt c releases into the cytosol where it forms an apoptosome, together with the cytosolic protein Apaf-1, dATP and procaspase 9. After proteolysis of procaspase 9, caspase 9 activates caspases 3, 6 and 7 that execute the downstream apoptotic processes such as cleavage of nuclear lamina, DNA fragmentation, etc. In addition to Cyt c, other proapoptotic IMS proteins: Smac/DIABLO, Omi/HtrA2, endonuclease G, and apoptosis-inducing factor (AIF) are released from the injured mitochondria. Smac/DIABLO neutralizes the apoptosis inhibitor protein (IAP), and thus accelerates the caspase-dependent apoptotic pathway. Endonuclease G and AIF participate in the fragmentation of DNA, and subsequent chromatin condensation, independent of caspases (Figure 3).
There are two possible mechanisms for the release of Cyt c and other IMS proteins from mitochondria:
(i) Opening of the mitochondrial permeability transition pores (PTP) is a key event in cell death. PTP is a protein complex in the contact site between the outer and inner mitochondrial membranes (OMM and IMM, respectively). It consists of: (1) the voltage-dependent anion channel (VDAC), integrated into OMM; (2) the translocator of adenine nucleotides (ANT), a component of IMM; (3) the peripheral benzodiazepine receptors (PBR) interacting with VDAC; (4) the matrix protein cyclophilin D that interacts with ANT. Opening of PTP causes an instant ion leakage, loss of ΔΨm, uncoupling of oxidative phosphorylation, inhibition of ATP synthesis, and hyperproduction of mROS. Massive water influx through opened PTP causes osmotic matrix swelling followed by rupture of OMM, whose surface is much smaller than that of IMM. This promotes release of stored Ca2+ and IMS proteins into cytosol.
(ii) Opening of specific megachannels in the OMM, large enough for protein passage, which are formed by homooligomers of Bcl-2 family proteins Bax or Bak. Bak normally resides in OMM, whereas Bax is present mostly in cytosol where it is associated with the DNA repair factor Ku70. In response to cell damage, this complex dissociates and liberates Bax. Bax is then translocated into OMM, where it oligomerizes and forms a megachannel for the passage of Cyt c and other proapoptotic proteins. The anti-apoptotic proteins, Bcl-2 and Bcl-xL form heteropolymers with Bax and Bak and thus reduce their fraction involved in the channel formation. So called "BH-3 only proteins", such as Bid and Bim, bind to Bcl-2 and Bcl-xL and prevent their association with Bax and Bak, thus increasing the pro-apoptotic potential (Figure 3).
As shown in numerous publications, PDT rapidly induces mROS generation, ΔΨm dissipation, release of Cyt c, AIF and Smac/DIABLO, and following apoptosis in different cells sensitized by various photosensitizers (PS). In the case of photosensitizers localized to mitochondria such as protoporphyrin IX, intensely generated 1O2 and mROS cause peroxidation of highly unsaturated cardiolipin, which anchors Cyt c. Following mobilization and release, Cyt c activates caspases 3, 6, 7, and the downstream apoptotic processes. Both ways of Cyt c release from mitochondria have been demonstrated in photosensitized cells: through the opened PTP and through the Bax/Bak megachannels.
PDT can either open or, oppositely, inhibit PTP. Some porphyrins bound specifically to PBR inactivate PTP, and prevent its opening. In contrast, the involvement of PBR, a PTP component in PDT-induced apoptosis, has been also shown (Kessel et al., 2001).
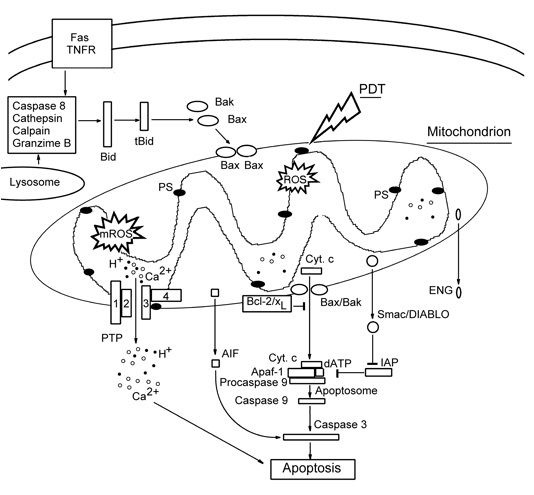
Figure 3. Mitochondria-controlled apoptosis pathways.
AIF: apoptosis-inducing factor, ENG: endonuclease G, Fas and TNFR: apoptosis-inducing membrane receptors, IAP: apoptosis inhibitor protein.
In some photosensitized cells, Cyt c release and the resultant apoptosis occurred independently of PTP opening and ΔΨm loss. These processes were controlled by the Bcl-2 superfamily proteins: Bcl-2, Bcl-xL, Bax, and Bak.
Overexpression of anti-apoptotic proteins Bcl-2 and Bcl-xL usually delays or prevents PDT-induced Cyt c release, activation of caspases 3, 6 and 7, and apoptotic DNA fragmentation (Granville et al., 1999; Vantieghem et al., 2001). On the contrary, the decrease of the Bcl-2 level caused, for example by antisense Bcl-2 RNA, promotes apoptosis despite the unaltered level of proapoptotic protein Bax (Srivastava et al., 2001). Unexpectedly, overexpression of Bcl-2 enhanced apoptosis in some photosensitized cells. However, in these cells the pro-apoptotic protein Bax was simultaneously up-regulated, so that the Bax/Bcl-2 ratio increased (Kim et al., 1999). Therefore, the cell fate is determined by the balance between Bax and Bcl-2 levels. The antiapoptotic effect of Bcl-2 and Bcl-xL is probably related to their ability to form heterodimers with Bax and thereby decrease the pool of Bax molecules, which form the megachannels for passage of pro-apoptotic proteins through OMM (Bras et al., 2005). This explains why the increase in the Bax/Bcl-2 ratio may enhance the pro-apoptotic potential. In photosensitized cells, Bax level may be increased, or decreased, or unaltered, but the Bax/Bcl-2 ratio is elevated in cells committed to apoptosis.
Bax is also involved in the release of other pro-apoptotic proteins such as AIF or Smac/DIABLO from mitochondria to cytosol in photosensitized cells (Usuda et al., 2002).
4. Apoptosis Triggered by Photodynamic Targeting of the Plasma Membrane or Lysosomes
Although PM-localizing photosensitizers generally induce necrosis, they can sometimes induce apoptosis through the activation of Fas, the surface proapoptotic receptor, and downstream caspase 8, which further stimulates caspases 3, 6 and 7. The increased expression of Fas and Fas ligand after the PDT treatment of mouse tumors could be due to intercellular interactions. However, Pc 4-PDT also increased the expression of Fas, Fas ligand, and FADD (an adapter molecule for Fas), as well as Fas multimerization and Fas binding to FADD in cultured cells, where intercellular interactions were limited (Ahmad et al., 2000). Caspase 8 activation in photosensitized cells was accompanied by cytosolic release of Cyt c and AIF (Takahashi et al., 2003).
Photosensitizers localizing in lysosomes may induce cell death with features of necrosis, or apoptosis, or their combination. The rapid disruption of lysosomes in response to the photodynamic effect of hydrophilic PS may cause the release of cathepsin, which cleaves cytosolic protein Bid. The truncated protein tBid induces Bak oligomerization and the formation of pores in OMM, through which Cyt c releases from mitochondria and initiates caspase 3-mediated apoptosis (Figure 3) (Reiners et al., 2002).
5. The Role of Ca2+ in Photodynamic Cell Injury
5.1. PDT effect on cellular Ca2+ homeostasis. Intracellular 1O2 concentration, [Ca2+]i, is kept normally at a very low level: 10-8-10-7 M. There are several pathways that increase the cytosolic Ca2+ level (Figure 4). Ca2+ may penetrate through potential-dependent or receptor-operated channels in the plasma membrane. It may be also exchanged by intracellular Na+. Ca2+ is also released from intracellular Ca2+-storing organelles: mitochondria or ER. The release of Ca2+ stored in mitochondria occurs after PTP opening or IMM disruption. Ca2+ may also be exchanged by cytosolic H+ or Na+. Ca2+ release from ER occurs through channels formed by inositol 1,4,5-phosphate or ryanodine receptors. Small amount of cytosolic Ca2+ may induce massive Ca2+ release from ER (Ca2+-induced Ca2+ release, CICR). Likewise, there are several pathways for decreasing the intracellular Ca2+ level, [Ca2+]i. Ca2+ may be pumped out by the plasma membrane Ca2+-ATPase (PMCA, or sequestered into ER by Ca2+-ATPase (SERCA). It is also accumulated in mitochondria by the mitochondrial Ca2+ uniporter (Figure 4).
Ca2+ serves as a second messenger in the signal transduction system. It integrates different signaling pathways (Figures 1, 5), and regulates numerous cellular processes. Sustained [Ca2+]i elevation may induce both necrosis and apoptosis. Ca2+ activates various proteinases, nucleases and phospholipases. It induces PTP opening with the resultant Cyt c release from mitochondria, and activation of a caspase cascade. Ca2+-activated signaling pathways involving JNK, transcription factors ATF and ELK, calcineurin (protein phosphatase 2B) and apoptosis associated tyrosine kinase (AATyK) may also induce apoptosis. On the other hand, Ca2+-related pathways involving NF-kB and calmodulin/kinases II or IV usually support cell survival (Figure 5).
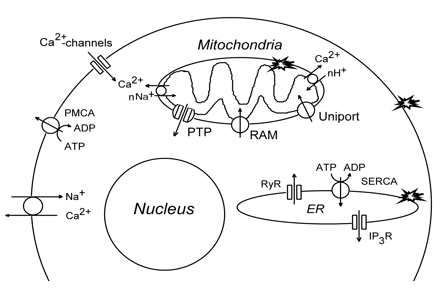
Figure 4. The intracellular systems maintaining Ca2+. IP3R: inositol-1,4,5-trisphosphate receptor, PMCA: plasma membrane Ca2+-ATPase, PTP: permeability transition pore, RAM: rapid accumulation of Ca2+ in mitochondria, RyR: ryanodine receptor, SERCA: ER Ca2+-ATPase. Stars show PDT targets.
PDT rapidly increases [Ca2+]i in diverse cell lines. A transient [Ca2+]i rise is generally followed by sustained elevation. PDT-induced [Ca2+]i increase depends on subcellular PS localization. Hydrophobic PSs release Ca2+ mainly from the photodamaged stores, mitochondria and/or ER. Hydrophilic PSs adsorbed at PM induce Ca2++ influx through ion leak pathways and PM ruptures. Additionally, [Ca2+]i increase may result from the photodamage of calcium pumps or the activation of phospholipases and the resultant breakdown of membrane phosphoinositides. The transient [Ca2+]i increase is suggested to be a result of ion influx through PM, whereas the photoinjury of Ca2+-storing organelles may be responsible for prolonged Ca2+ accumulation. A comparison of microsomes and mitochondria showed that Ca2+ influx into ER was more impaired by hematoporphyrin- or protoporphyrin-mediated PDT than was Ca2+ influx into mitochondria. Ca2+-transporting enzymes, SERCA and Ca2+ uniporter, were suggested to be the preferential targets for PDT (Ricchelli et al., 1999). The Golgi-specific photosensitizer, TBR, sensitized Ca2+ release from the Golgi apparatus, and induced caspase-independent apoptosis (Ogata et al., 2003).
The antiapoptotic protein Bcl-2 that is found in OMM, ER and the nuclear envelope regulates [Ca2+]i in photosensitized cells. Cells overexpressing Bcl-2 accumulate more Ca2+ in mitochondria and generate greater [Ca2+]i transients in response to PDT. PDT-induced degradation of SERCA leads to [Ca2+]i increase. Ca2+ may activate caspases and endonucleases that execute apoptotic fragmentation of DNA (Granville et al., 2001).
Mitochondria accumulate large amounts of calcium, which may be released upon injury. PDT differently influences this processes in different conditions. mTHPC-PDT has been shown to inhibit Ca2+ uptake by isolated mitochondria due to injury of Ca2+-uniporter (Klein et al., 1997). Hematoporphyrin-PDT inactivated ANT in isolated mitochondria and prevented PTP opening. These mitochondria, however, retained ΔΨm and the ability to accumulate Ca2+ (Salet et al., 1997). Sensitizers localized to different sites than PBR, which binds porphyrins specifically, induced PTP opening. It was surprising that PDT-induced PTP opening did not impair mitochondrial electron transfer and oxidative phosphorylation (Moreno et al., 2001). Therefore, PDT may act differently on Ca2+ homeostasis depending on PS localization.
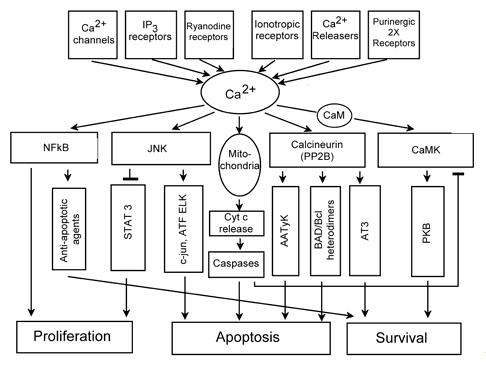
Figure 5. Ca2+-related signaling processes in cells.
AATyK: apoptosis associated tyrosine kinase, ATF, ELK, AT3: transcription factors, CaMKII: calmodulin-dependent protein kinase, IP3: inositol-1,4,5-trisphosphate, JNK: c-Jun terminal kinase, NF-κB: nuclear factor-κB, PKB: protein kinase B, STAT: signal transducer and activator of transcription (Modified from Anis et al., 2006).
5.2. The role Ca2+ in PDT-induced cell death. There are different opinions on the role of cytosolic Ca2+ in photosensitized cells. The majority of authors showed that PDT-induced elevation in [Ca2+]i leads to cell death. It may occur through either caspase-dependent or caspase-independent apoptosis. This is related to Ca2+-mediated activation of various hydrolytic enzymes. For example, in photosensitized HL60 cells, Ca2+ activated protease calpain that triggered the caspase 3-mediated apoptotic cascade. Simultaneously, Ca2+ activated endonuclease G that cleaved DNA independently of caspase 3 (Grebenova et al., 2003). In HeLa cells, photosensitized with verteporfin, photodegradation of the calcium pump SERCA2 increased [Ca2+]I, which activated protein phosphatase 2B calcineurin, and induced calcineurin-mediated dephosphorylation of Bad, inactivation of Bcl-xL, and subsequently apoptosis (Granville et al., 2001). Elevation of [Ca2+]i after photodynamic injury of SERCA2 by ER-localizing hypericin induced rapid Bax/Bak-dependent apoptosis with Cyt c release, ΔΨm loss, caspase 3 and PARP cleavage. Cells lacking Bax and Bak died from caspase-independent autophagy (Buytaert et al., 2006). However, some authors indicate Ca2+ involvement in cell protection (Ben-Hur and Dubbelman, 1993).
PDT-induced [Ca2+]i elevation modulated various cell functions, such as muscle contraction or neuronal activity. For example, photosensitization of an isolated neuron with sulphonated aluminium phthalocyanine caused the gradual inhibition and subsequent irreversible abolition of neuronal activity. Application of different modulators of Ca2+ channels, SERCA inhibitors, or Ca2+-ionophore showed that firing inhibition was associated with cytosolic Ca2+, independent of its source. Ultrastructural studies revealed mitochondria and ER injury in photosensitized neurons, which corresponded to their involvement in [Ca2+]i increase (Uzdensky et al., 2000; 2002, 2007).
6. The Role of Ca2+-Related Signaling Pathways in the Effect of PDT on Cells.
6.1. Phospholipases A2 and C. Phospholipase-mediated degradation of specific phospholipids generates diverse second messenger products. Phospholipase A2 (PLA2) may be activated by Ca2+, protein kinase C, and MAPK. It generates arachidonic acid, a substrate for the production of paracrine regulators such as prostaglandins and tromboxanes. Phospholipase C (PLC) is regulated by extracellular molecular signals recognized by RCGP or RTK. It produces two second messengers: IP3 that stimulates Ca2+ release from ER, and diacylglycerol that activates protein kinase C. Phospholipases also destroy membrane lipids, impair PM functions, cause ion leakage, and finally necrosis.
It was shown that Pc 4-PDT rapidly activated PLC in mouse lymphoma cells and transiently elevated IP3 level that was followed by Ca2+ release from ER. Ca2+ in turn activated PLA2 to produce arachidonic acid that was involved in subsequent apoptosis (Agarval et al., 1993). The situation is, however, not so simple, because prostaglandin E2 produced from arachidonic acid could simultaneously initiate cell protection pathways as shown in HpD-sensitized human carcinoma T24 cells (Penning et al., 1993b).
6.2. Calmodulin and calcium/calmodulin-dependent protein
kinase II. Calmodulin (CaM), an abundant cellular protein, is activated by Ca2+ to control numerous cellular processes including cyclic nucleotide metabolism, ion transport, cytoskeleton remodeling, activation of diverse protein kinases and phosphatases (Figure 4). It is considered as a "decoder" of calcium information in the cell. CaM-dependent protein kinases (CaMK I, II, or IV) are activated by calmodulin in response to [Ca2+]i increase. They regulate numerous cellular functions and especially expressed in brain.
Despite the significant photodynamic alteration of Ca2+ levels, the role of CaM and CaMK in the cellular response to PDT remains practically unstudied. Grebenova et al. (2003) has reported that ALA-PDT-induced an elevation of CaM levels in HL60 cells. These authors suggested CaM involvement in cell protection. The involvement of CaMKII and CaMKIV in protection of breast cancer cells from oxidative stress induced by ionizing radiation, H2O2, or PDT has been recently demonstrated. Oxidative stress increased CaMK activity, whereas CaMK inhibition enhanced cell killing (Rodriguez-Mora et al., 2006). In the photosensitized crayfish stretch receptor, both CaM and CaMKII were involved in PDT-induced necrosis of neurons and glial cells, but not in glial apoptosis (Uzdensky et al., 2007).
6.3. Protein kinase C. Protein kinase C is one of the central regulators in the cell signaling system. After activation by Ca2+ and diacylglycerol, it phosphorylates and regulates various proteins controlling cell life and death including numerous enzymes, ion channels, signaling proteins, transcription factors, and cytoskeletal proteins.
The literature on the role of PKC in PDT-induced cell death are contradictory, probably due to its involvement in different signaling pathways. For instance, PKC was shown to be involved in the PDT-induced loss of the clonogenic activity of CHO cells sensitized with alumophthalocyanine; cell killing was enhanced by phorbol ester TPA, an activator of PKC, whereas PKC inhibition by H7 protected these cells (Rash et al., 1997). PKC was also involved in PDT-induced inhibition and abolition of firing in photosensitized crayfish neurons and in apoptosis of glial cells (Bragin et al., 2003; Uzdensky et al., 2007).
Other data, in contrast, demonstrate the protective, anti-apoptotic role of PKC in different cell lines. PKC inhibitors: calphostin C, hypericin, staurosporine, or tamoxifen facilitated PDT-induced apoptosis. Some of them such as hypericin and calphostine C are photosensitive. Light exposure sharply increased their ability to inhibit PKC, their anti-proliferative and pro-apoptotic activity. These drugs simultaneously acted as photosensitizers and PKC inhibitors. It is of interest that malignant glioma cells, like normal neonatal cells, but not mature glial cells, express high levels of PKC. Its photoinhibition mediated by hypericin or calphostin C may provide a strategy for the selective blocking of tumor growth (Pollack, Kawecki, 1997; Weller et al., 1997).
The effector proteins activated by PKC in photosensitized cells, and the downstream processes are unidentified. Only fragmentary data have been obtained. For example, PKC is known to phosphorylate and activate Bcl-2 that is involved in apoptosis prevention (Zhuang et al., 1998). PKC also participates in PDT-induced activation of the early response gene c-fos (Luna et al.,1994), and transcription factor NF-κB that may contribute to cell survival (Volanti et al., 2005).
7. Cyclic AMP-Dependent Signaling Pathway
Cyclic AMP (cAmp) is produced by adenylate cyclase, which is stimulated by G-protein in response to binding of an extracellular signaling molecule (hormone, neuromediator) to RCGP. Then cAMP activates protein kinase A, which regulates numerous cellular processes (Figure 1). This pathway is commonly involved in cell protection.
The literature generally confirms the protective role of cAMP in PDT-treated cells. For example, photosensitization of carcinoma cells with hematoporphyrin derivative transiently increased the intracellular cAMP level. The permeable cAMP analog, 8'-bromo-cyclic AMP, AC activator forskolin, prostaglandin E2, which stimulates RCGP, all reduced phototoxicity (Penning et al., 1993a). AC activation by forskolin prevented PDT-induced apoptosis in V79 cells. Forskolin inhibited caspase-3 activation, but not Cyt c release from mitochondria. Therefore, cAMP inhibited the apoptotic pathway between Cyt c release and caspase-3 activation (Inanami et al., 1999).
The similar protective action of the cAMP-mediated signaling pathway has been observed in neuronal and glial cells. AC and PKA were shown to be involved in the maintaining of neuronal activity that was impaired by PDT and protection of glial cells from PDT-induced apoptosis. PKA also protected neurons from PDT-induced necrosis. However, AC was, surprisingly, involved in PDT-induced necrosis of neurons and glial cells (Uzdensky et al., 2005; 2007). Therefore, the role of the cAMP-mediated signaling pathway may be more complicated. Its interactions with other signaling pathways, as well as the downstream effector processes should be studied more carefully.
8. Signaling Pathways Initiated by Receptor Tyrosine Kinases
8.1. Receptor tyrosine kinases. Receptor tyrosine kinases responding to cytokines and growth factors initiate three important signaling pathways mediated by phospholipase C, MAP kinases, and PI 3-kinase, which manage cell response to stress, control cell survival, and maintain tissue integrity (Figure 1). PDT may injure cell surface receptors. After adsorption on the cellular surface or dissolving in the lipid bilayer, photosensitizer molecules land in the vicinity of receptor proteins, and may affect them under light exposure. As a result, intercellular signaling may be significantly impaired.
The following PDT-mediated reactions were described (Liu et al., 2004): (1) structural modifications and loss of EGF receptors; (2) specific cross-linking of some proteins, namely STAT (signal transducer and activator of transcription); (3) dephosphorylation of tyrosine-phosphorylated proteins; (4) inhibition of protein phosphatases; (5) stimulation of JNK-related signaling pathway. This caused cell insensitivity to inflammatory cytokines. As demonstrated by Glinski et al. (1995), photosensitization of different cell lines significantly inhibited the binding of cytokines (TNF-α, IL-8, complement factor 5a, EGF) to their receptors, as well as the binding of monoclonal antibodies to cell surface antigens. This photoinhibition was not receptor-, cytokine-, or antibody-specific.
Hypericin-PDT strongly and irreversibly inhibited the tyrosine kinase activity of EGF and insulin receptors in crude preparations of cellular membranes (Agostinis et al., 1996). Rose Bengal-PDT, which targets PM, inhibited the tyrosine kinase activity of platelet-derived growth factor (PDGF) receptors in murine fibroblasts. The dimerization of PDGF receptors was observed, but their autophosphorylation or the phosphorylation of their substrate SH-PTP2 did not occur, possibly due to protein cross-linking in these dimers (Zhuang et al., 2003). ALA- or Photofrin-mediated PDT caused degradation of EGF and interleukin-6 receptors in different normal or cancerous epithelial cell lines (Wong et al., 2003).
Therefore, PDT makes cells refractory to extracellular signals, impairs cellular functions and leads to cell death.
8.2. Mitogen-activated protein kinase-related pathways. Mitogen-activated protein kinase (MAPK) pathways are involved in cell responses to various chemical or physical stimuli such as hormones, cytokines, oxidative stress, UV and ionizing radiation. MAPKs compose a family of conservative Ser/Thr kinases. They regulate gene expression, metabolism, movement, division, and cell death (Johnson and Lapadat, 2002).
There are three well-characterized MAPKs known: ERK, JNK/SAPK, and p38 (Figure 1). Two known ERK isoforms, ERK1 and ERK2, are stimulated by growth factors, cytokines or other chemical signals. Their downstream targets include other protein kinases such as MAPKAP-K1, MNK, or MSK, and transcription factors such as Elk-1. JNK/SAPK and p38 are activated in response to environmental stress caused by ionizing radiation or reactive oxygen species, as well as in response to growth factors and inflammatory cytokines (Figure 2). The JNK/SAPK family is subdivided by JNK1, JNK2 and JNK3. They phosphorylate the protein, c-Jun, which together with c-Fos forms the transcription factor AP-1, which controls many genes involved in cell response to stress. JNK targets such transcription factors as ATF2 and Elk-1. It plays an important role in cancer development and apoptosis. The p38 family consists of four isoforms: p38α, p38β, p38γ, and p38δ. Several transcription factors such as ATF-2, Elk-1, MAX, and CREB as well as protein kinases MSK, MAPKAP-K2 are phosphorylated by p38. JNK and p38 are phosphorylated and activated by upstream kinases: MAPKK (MEK 3, 4, 6 and 7) and MAPKKK (ASK1 and TRAF2) (Fig. 2). ASK1 may possibly serve as a ROS sensor reacting to oxidative stress and initiating MAPK signaling pathways, as indicated above.
8.2.1. Extracellularly regulated kinase. The data on the role of extracellularly regulated kinase (ERK) in cell responses to PDT are contradictory. Some authors did not observe any changes in ERK activity in photosensitized cells, whereas others reported ERK inhibition. In some cases, PDT activated ERK. For example, Tong et al (2002) have reported a rapid ERK activation in PDT-sensitive and PDT-resistant cell lines photosensitized with Photofrin. In PDT-sensitive cells, ERK activation was transient, whereas in PDT-resistant cells, it lasted more than 11 h. The simultaneous expression of MAP-activated phosphatase (MKP-1), which dephosphorylates ERK, implied its involvement in the regulation of the duration of ERK activation. It was shown that MEK kinase was the upstream activator of ERK in photosensitized cells. The combined action of ERK inhibitor and Photofrin-PDT significantly augmented the photodynamic killing of PDT-resistant cells, which suggested the protection role of the MEK/ERK cascade against Photofrin-PDT.
It is unclear so far, how PDT-produced ROS activate the MEK/ERK signaling pathway. The downstream ERK substrates in PDT-treated cells are also not established.
8.2.2. JNK and p38. As shown by many authors, PDT causes the rapid and significant time- and dose-dependent activation of JNK and p38 in different cell lines. The basal level of these MAP kinases could not change, but the level of their phosphorylated/activated forms rapidly increased within 30 min after PDT. Then the enzyme activity returned to the basal level or remained elevated (Zhuang et al., 2000; Hsien et al., 2002).
The role of JNK and p38 in PDT-induced cell dearth is also controversial. Some authors have reported that photoinduced activation of JNK or p38 leads to apoptosis, whereas others indicate their protective role. This could be due to a difference in MAPK-related signaling pathways in different cell lines. In A431 cells photosensitized with Rose Bengal, fast JNK activation was required for the activation of caspase 3 and the resultant apoptosis. Caspase 3 also cleaved/activated p21-activated kinase 2 (PAK2), which in turn activated JNK. This feedback loop caused a sustained late-stage activation of JNK, which exacerbated apoptosis (Chan et al., 2000). In HL-60 cells, Rose Bengal-PDT activated p38, caspase 3 and the downstream apoptotic cascade. Authors found that activation of Bid, a Bcl-2 family protein, linked the PDT-induced phosphorylation/activation of p38 with the release of Cyt c, activation of caspase 3 and other executioner caspases that perform apoptosis (Zhuang et al., 2000).
Hsien et al. (2002) observed rapid activation of JNK by Photofrin-PDT in A431 cells. This was followed by activation of caspase-3, PARP, and loss of ΔΨm characteristic for apoptosis. However, the morphological changes in these cells indicated necrosis. Therefore, initiated apoptotic core program might be not completed in these cells, and necrosis became dominant after PM injury. In lymphoma Ly-R cells constitutively expressing p38, PC 4-PDT significantly activated JNK but not ERK and p38. In CHO cells, which do not normally express p38, all MAPKs, ERK2, p38 and JNK, were activated by PDT. Inhibition of p38 led to the inhibition of caspases 9 and 3 and apoptosis suppression. Therefore, p38 acted upstream of this caspase cascade. JNK could also contribute to PDT-induced apoptosis (Xue et al., 1999b). In contrast, the strong and sustained activation of JNK1 and p38 that occurred without changes in their basal levels played a protective role in hypericin-sensitized HeLa cells (Assefa et al., 1999). As shown by these authors, hypericin-PDT inhibited the p38-dependent up-regulation of cyclooxygenase-2, and the resulting secretion of prostaglandin PGE2 in HeLa cells. This was presumably due to p38-mediated stabilization, but not transcription of the cyclooxygenase-2 mRNA (Hendrickx et al., 2003).
Thus, the role of MAPK in the cellular response to photodynamic treatment depended significantly on the cell type and the sensitizer used. For example, in cells photosensitized with highly lipophilic hypericin, which specifically localizes to ER and Golgi apparatus, activation of JNK and p38 was directed to the protection of cells from photoinduced apoptosis (Assefa et al., 1999; Hendrickx et al., 2003). In contrast, activation of these MAPKs in cells photosensitized with hydrophilic Bengal Rose or Photofrin localized to PM and lysosomes induced apoptosis (Chan et al., 2000; Zhuang et al. 2000). This is consistent with the growing experimental evidence that in different, context-specific situations, JNK may be required either for cell survival of for its death.
How 1O2 and other ROS induce JNK and p38, and which downstream processes activated by these MAPKs lead to cell death or survival are among unresolved problems.
8.3. PI 3-kinase/Akt-related pathway. Phosphatidylinositol 3-kinase (PI 3-kinase) is involved in cell responses to stress impacts, and regulates cell survival and resistance to apoptosis through the activation of protein kinase B/Akt (PKB/Akt pathway). There are several works in the literature on the possible involvement of PI 3-kinase and Akt in PDT-induced cell death.
Xue et al. (1999a) have demonstrated the involvement PI 3-kinase in the protection of prostate carcinoma cells from apoptosis induced by Pc 4-PDT. Etk/Bmx tyrosine kinase was found to be an effector of PI 3-kinase in these cells. Inhibition of PI 3-kinase abolished Etk activity and enhanced apoptosis. Photosensitization of murine fibroblasts with Rose Bengal-PDT caused sustained activation of Akt and p38 that was followed by apoptosis. Activation of p38 was suggested to be a death signal, while Akt activation is a survival signal. Akt was activated by PI 3-kinase, whose inhibitors wortmannin or LY294002 prevented photoactivation of Akt and enhanced cell death, indicating Akt involvement in cell survival (Zhuang and Kochevar, 2003). PDT treatment reduced the activation of Akt that was caused by growth factors EGF or PDGF. Two parallel but oppositely directed pathways of Akt regulation were suggested: (i) Activation of PI 3-kinase/Akt pathway and (ii) Production of ceramide that may stimulate phosphatases PP1 and PP2 to inhibit Akt and ERK. The Akt effect on cell survival could be mediated by phosphorylation and thus inhibition of glycogen synthase kinase 3 (Schieke et al., 2004).
PI 3-kinase has been recently shown to participate in the formation of autophagosomes in hypericin-sensitized Bax-/Bat- double knockout cells that died from PDT-induced autophagy (Buytaert et al., 2006).
In the electrophysiological experiments on crayfish mechanoreceptors, inhibitors of PI 3-kinase slowed down the PDT-induced inhibition of neuronal activity and delayed neuron inactivation. Since photoinduced firing inhibition is known to be associated with [Ca2+]i increase, it was suggested that PI 3-kinase and Ca2+ signaling pathways interacted in mediating the neuron response to PDT (Bragin et al., 2003). In fact, PI 3-kinase is known to stimulate Ca2+ influx into diverse cell types. On the other hand, Ca2+/CaM may activate PI 3-kinase. This illustrates the importance of cross-talk between different signaling processes in the integral cellular response. Protein kinase C activated by Ca2+ was found to be among PI 3-kinase targets in photosensitized carcinoma T24 cells, but not in epithelial carcinoma HeLa cells (Volanti et al., 2005).
9. Protein Phosphatases
Protein phosphatases are very important for cell signaling. They dephosphorylate proteins and thus prevent their activation. This is necessary for the temporary organization of signaling processes. Their role in cellular effects of PDT is not well studied. The dual specificity phosphatase 1 was among 40 from 2800 studied proteins, whose mRNA was up-regulated in normal and tumor urothelial cell lines after ALA-PDT (Wild et al., 2005). Photofrin-PDT induced expression of MAPK phosphatase MKP-1, which regulated the duration of activation of ERK, and was involved in cell protection (Tong et al., 2002). In the crayfish stretch receptor, inhibition of protein tyrosine phosphatase by sodium orthovanadate delayed PDT-induced inhibition of neuronal activity and reduced necrosis in satellite glial cells. This showed the involvement of tyrosine phosphatase in the photoinactivation of neurons, and the necrosis of glial cells (Uzdensky et al., 2005).
10. Transcription Factors and the Early Response Genes
The key protein kinases activate various transcription factors, and thus regulate the expression of genes that produce proteins involved in integral cell reactions. AP-1 and NF-κB are the best characterized transcription factors involved in diverse physiological functions including, cell response to stress.
10.1. AP-1. The transcription factor AP-1 (activating protein-1) controls the expression of diverse genes involved in apoptosis, proliferation, differentiation, tumor invasion, angiogenesis, and other cellular functions. It is a homo- or heterodimeric protein formed from proteins of the Fos and Jun families. Such combinations provide a variety of gene expression patterns. AP-1 can be activated by growth factors, cytokines, hypoxia, ionizing and UV radiation. It is a redox-sensitive transcription factor, but the oxygen sensing mechanism is unclear. Ca2+-related pathways also modulate AP-1 signaling.
PDT rapidly induces the expression of the early response genes, c-fos and c-jun, and AP-1 binding to DNA in different cancer cells. Furthermore, PDT increased the stability of c-jun and c-foc mRNA (Luna et al., 1994; Wild et al., 2005). The inhibition of phospholipase PLA2 or protein kinases C inhibited PDT-mediated activation of c-fos. However, inhibitors of calmodulin, protein kinase A, or phosphodiesterase had no effect. Therefore, the PDT-induced activation of c-jun, c-foc and AP-1 was mediated by PKC and PLA2, but not by cAMP-dependent signaling pathway (Luna et al., 1994).
10.2. NF-κB. The transcription factor NF-κB serves as a central integrator of stress responses and cell survival pathways. It controls transcription of over 150 target genes. NF-κB is involved in the inhibition of apoptosis, stimulation of cell proliferation, inflammation, immune response, and tumorigenesis. It is conserved from Drosophila to man. NF-κB is commonly the p50/RelA heterodimer. It binds to the specific consensus DNA site. In the cytoplasm, NF-κB forms an inactive complex with an inhibitory protein IκB, which covers its nuclear localization fragment, thereby keeping it in the cytoplasm. NF-κB activity is regulated by the formation and decay of the IκB–NF-κB complex. Cells contain a specific IκB kinase complex (IKK). When stimulated, IKK phosphorylates IκB, which induces decay of the IκB-NF-κB complex, with the resulting proteasome-mediated degradation of IκB. Free NF-κB is rapidly translocated into the nucleus, where it binds to DNA and promotes the transcription of certain genes. One of them encodes IκBα, an IKK component, which enters the nucleus, binds to NF-κB, and returns back to the cytosol as an inactive complex.
NF-κB can be activated by a variety of stimuli including cytokines, growth factors, hormones, hypoxia, ionizing and UV radiation, and PDT. Diverse physiological processes such as depolarization, neuronal activity, adhesion, or hypoxia also induce NF-κB. Most of these factors produce ROS and cause oxidative stress. That is why NF-κB is known as a redox-activated transcription factor, which plays a significant role in cellular response to PDT. However, the signaling pathways leading from intracellular ROS targets to IKK are unknown so far. Activation of NF-κB generally prevents apoptosis. NF-κB up-regulates anti-apoptotic genes encoding Bcl-2, Bcl-xL, c-Myc, survivin, cIAP 1/2, XIAP, COX-2, MnSOD and other cell protection proteins, and down-regulates some pro-apoptotic proteins (Matroule and Piette, 2000; Aggarwal et al., 2006).
PDT treatment with various PSs activated NF-κB in diverse cell lines. This was evident from the disappearance of IκB in the cytoplasm, and the appearance of NF-κB heterodimer p50/p65 in the nucleus. PDT also enhanced NF-κB binding to DNA (Ryter, Gomer, 1993).
Verteporphyn-PDT that activated NF-κB, also rapidly induced apoptosis mediated by Cyt c, caspases 3 and 9 (Granville et al., 2000). However, in HCT-120 cells photosensitized with PMME, these events were independent. PMME localizes predominately to ER and Golgi, but not mitochondria. Therefore, authors suggested that primary 1O2-generation was followed by the secondary generation of diffusible ROS, such as O2.- and H2O2, which could induce the burst generation of mROS and apoptosis. The mechanism of PDT-induced activation of NF-κB did not depend on ROS generation, but required internalization of interleukin IL-1 and the recruitment of the signaling machinery, in which IKK phosphorylates IκBα and thus promotes its degradation. Apoptosis induction was independent on IL-1. Although NF-κB played a protective role in these cells, the anti-apoptotic genes remain to be identified (Matroule et al., 2001). PMME-PDT caused a sustained activation of NF-κB, and the expression of NF-κB-controlled genes in endothelial cells. This process was associated with IKK-independent degradation of the inhibitory protein IκBα. The activation of IKK and NF-κB in T24 cells was mediated by PI 3-kinase and PKCα (Volanti et al., 2002; 2005).
10.3. HIF-1. The hypoxia-inducible factor (HIF-1) is a ubiquitous transcription factor involved in the control of cell and tissue responses to hypoxia, specifically in angiogenesis, hematopoiesis and anaerobic energy metabolism. It may serve as a cellular oxygen sensor.
PDT induces rapid oxygen consumption and a decrease in the intracellular oxygen level. PDT-mediated microvascular injury additionally increases tumor hypoxia (Moan and Juzeniene, 2008). Koukourakis et al. (2001) reported the expression of HIF1α and HIF2α proteins in PDT-treated esophageal cancer. The high expression of HIF was associated with a low rate of complete tumor response, and with the absence of Bcl-2 protein expression. On the contrary, Bcl-2 expression was associated with a high complete response rate. Therefore, HIF and Bcl-2 expression may predict tumor sensitivity to PDT.
Esophageal Het-1A cells, in which high expression of HIF-1α was pre-induced by CoCl2-mediated chemical hypoxia, were more resistant to ALA-PDT than cells without cobalt treatment. The transfection of these cells with anti-HIF-1α short interfering RNA prevented HIF-1α expression and restored cell photosensitivity. However, in esophageal tumor, CoCl2 did not induce the overexpression of HIF-1α and survival after ALA-PDT. Therefore, HIF-1α is of importance for the resistance of normal, but not cancerous esophageal cells to ALA-PDT (Ji et al., 2006).
10.4. p53 and related proteins.
p53. The tumor suppressor protein p53 coordinates DNA repair, cell cycle progression and DNA damage-dependent apoptosis. As a transcription factor, p53 regulates at least 100 genes participating in these processes. The essential physiological function of p53 is to destroy selectively stressed cells through apoptosis. Loss or mutation of p53 leads to tumor progression. p53 plays a central role in the cellular response to oxidative stress. Its level in normal cells is usually kept low due to fast proteolysis. Following extensive DNA damage induced by ROS, ionizing or UV radiation, the level of p53 protein is transiently increased. ROS-induced activation of p53 may be mediated by diverse signaling pathways: (i) cdk4/6 and pRb; (ii) ATM; (iii) JNK/p38; (iv) p21ras; and (v) Ca2+/calpain (Fig.6). The JNK pathway is suggested to play the central role in p53 activation after oxidative stress. The downstream targets of p53 include genes the encoding pro-apoptotic proteins Fas, Apaf-1, Bax, PUMA and NOXA. The pro-apoptotic effect of p53 is supplemented with its inhibitory effect on the survival pathways mediated by such transcription factors as NF-kB and CREB. Thus, cell fate is a result of the fine balance between pro- and anti-apoptotic signaling pathways (Yu, Zhang, 2005).
DNA damage is not characteristic for photodynamic cell injury since the most sensitizers do not localize to the nucleus. Nevertheless, p53 can participate in PDT-induced cell death. Some works show that activation of p53 enhances PDT-induced necrosis (Tong et al., 2000), whereas others indicate p53 involvement in PDT-induced apoptosis (Fisher et al., 1999; Gupta et al., 2003; Barberi-Heyob et al., 2004).
It should be remembered that about 50% of all cancers carry mutations in the p53 gene, which allows them to avoid apoptosis, and to proliferate in an uncontrolled fashion. Accordingly, many tumor cell lines studied in PDT experiments are p53 mutants. Unfortunately, pharmacological agents for the modulation of p53 activity are absent. In order to elucidate the role of p53 in the cellular response to PDT, most authors have compared cell lines that express this protein differently, or used wild-type cells and mutants in the p53 gene, or mutants and cells with restored p53 activity after transfection of the p53 gene.
The expression of p53 does not modulate directly the sensitivity of either the apoptosis-responsive or non-apoptosis-responsive tumor cells to Photofrin-PDT (Fisher et al., 1999). The comparison between photosensitization of human glioma cells carrying wild-type p53 and human carcinoma cells with mutated p53 showed, however, the involvement of p53 in PDT-induced apoptosis (Gupta et al., 2003). The weakness of such experiments is in comparison of different cell lines. According to Tong et al. (2000), LFS cells with mutated p53 showed higher resistance to Photofrin-PDT than normal human fibroblasts, which express wild-type p53. Adenovirus-mediated expression of wild-type p53 in LFS cells enhanced their photodynamic injury. Many LFS cells were accumulated in G2 phase and died from apoptosis. Normal fibroblasts with a high p53 level died, however, from necrosis. Lee et al., (2005) studied the effect of hypericin-PDT on two isogenic cell lines: osteosarcoma U2OS and U2OS+p53DD, which differed only in the expression of p53. These authors did not observe any influence of p53 on hypericin uptake, cell cycle arrest profile, induction of apoptosis, or overall cell death. The resistance of some mutant variants of HT29 adenocarcinoma cells to Photofrin-PDT correlated with a decrease in the expression of mutated p53 and Bax, and an increase in Bcl-2 and Hsp27 levels (Shen et al., 2005). The re-establishment of wild-type p53 in colon cancer cells, in which the p53 gene was mutated, enhanced PDT-induced apoptosis, but not necrosis (Barberi-Heyob et al., 2004).
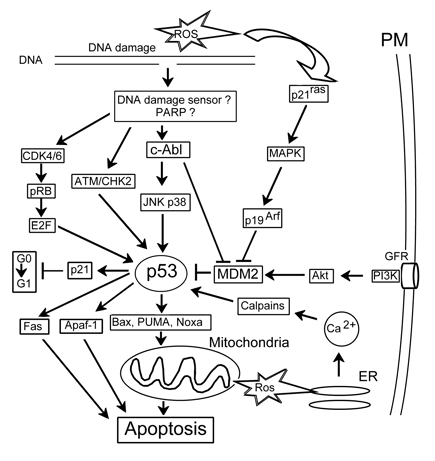
Figure 6. p53-related signaling. c-Abl: nonreceptor tyrosine kinase activated by DNA damage, APAF-1: apoptotic protease activating factor 1, ATM: protein kinase sensitive to DNA breaks, E2F: gene regulatory protein, GFR: growth factor receptor, pRB: retinoblastoma protein, PUMA and Noxa: proapoptotic members of the Bcl-2 family.
Photosensitization of mouse embryonic fibroblasts with m-THPC rapidly induced necrosis in most cells independent of proteins p53 and ATM, which transfer the information on DNA damage to p53. Some cells, however, died from apoptosis. The fraction of apoptotic cells was lower in fibroblasts that had lost p53 and ATM. Therefore p53 and ATM are required for PDT-mediated apoptosis, but not necrosis (Heinzelmann-Schwarz, 2003). There are two possible pathways to activate p53: (a) through activation of JNK and other signaling pathways, and (b) through the induction of multiple DNA breaks at the beginning of apoptosis. The activated p53 may induce the transcription of genes encoding pro-apoptotic proteins that remained undamaged. This can create a positive feedback loop accelerating DNA chopping and apoptosis progression. Both mechanisms could occur prior to significant p53 expression (Tong et al., 2000; Fisher et al., 1999). Thus, more data indicate t he involvement of p53 in cell responses to PDT than the data against this.
p21. The cyclin-dependent kinase inhibitor p21/WAF/Cip1 (named here as p21) causes cell cycle arrest. Following oxidative stress, p21 expression is induced by both p53-dependent and p53-independent mechanisms. As a proliferation inhibitor, p21 is involved in the prevention of tumor development. It also inhibits apoptotic processes and participates in DNA repair. Because of their central role in cell survival and death, p21 and p53 are considered master regulators of cell fate (Yu, Zhang, 2005).
The delayed expression of p21 and a block of G1 phase were observed in photosensitized human colon carcinoma cells (Fisher et al., 1999). Pc 4-PDT was shown to induce cell cycle arrest and apoptosis of A431 cells with a mutated p53 gene. Biochemical mechanisms of these processes included expression of p21 protein, down-regulation of cyclins D1 and E, and cyclin-dependent kinases cdk 2 and cdk6. This caused cell arrest in G0-G1 phase, and apoptosis of cells unable to repair the damage. Therefore, the overexpression of p21 may be related to pro-apoptotic rather than anti-apoptotic processes in PDT-treated cells. In cancer cells that lost p53 activity, p21 could deregulate the cell cycle and cause cell arrest in G0-G1 phase (Ahmad et al., 1998). However, the mechanism of p21 recruitment in PDT-induced apoptosis remains unclear.
10.5. Other transcription factors. Retinoblastoma (pRb) and E2F family transcription factors regulate the G1→S transition in the cell cycle. Ahmad et al., (1999) have demonstrated the involvement of pRb-E2F machinery in PDT-mediated cell cycle arrest and apoptosis. According to their hypothesis, Pc 4-PDT increases the production of NO., which in turn stimulates p21 to reduce the level of cyclins and cyclin-dependent kinases. This reduces the phosphorylation of pRb and down-regulates all five E2F family transcription factors. As a result, G1→S transition does not occur, and the cell cycle is arrested in G1 phase, which induces apoptosis. Fisher et al. (1999) observed a rapid transition of pRb from hyper- to hypophosphorylated state, which was not mediated by p53 or p21 expression in PDT-treated cancer cells that do not express p53.
Gene expression is regulated by signal transducer and activator of transcription proteins (STAT). Photodynamic therapy caused specific covalent cross-linking of STAT3, and to lesser extent of STAT1 and STAT4. Cross-linked STAT3 resided in the cytoplasm and failed to bind to DNA. As a consequence, photosensitized but survivivg cells lost their responsiveness to interleukin-6 and oncostatin M (Liu et al., 2004).
The role of other transcription factors such as CREB, ATF, etc., which participate in cell survival, apoptosis, or response to stress in photosensitized cells remains to be studied.
11. NO and NO Synthase
Nitric oxide (NO) plays the role of a second messenger in numerous physiological and pathological processes, including neurotransmission, vasodilatation, and cell response to stress. It is generated from L-arginine by NO synthase. NO activates guanylate cyclase to produce cyclic GMP (cGMP), which regulates diverse physiological processes.
PDT has been shown to induce NO generation in different cell lines. On the other hand, NO protects cells from PDT-induced apoptosis. It may trap cytotoxic free radicals and intercept lipid peroxidation in cellular membranes. As shown by Niziolek et al. (2003), the exogenous NO source spermine NONOate protected photosensitized cells from lipid peroxidation and subsequent necrosis. NO caused rapid (for 1-2 min) heme liberation and mobilization of free iron. This induced up-regulation of the protective proteins heme oxigenase-1 and ferritin for a much longer time (hours). Ferritin then reduced the free iron content below the initial level, thus providing cell resistance to the subsequent oxidative damage. Gomes et al., (2002) reported another mechanism of NO-mediated cell protection against PDT-induced apoptosis in lymphoblastoid cells photosensitized with AlPcS2 in the presence of the NOS substrate L-arginine or different exogenous NO donors (DETA NONOate, SNAP or hydroxylamine).
The protective effect of NO was eliminated by inhibitors of guanylate cyclase or cGMP-activated protein kinase G (PKG). The cGMP analog 8-Br-cGMP mimicked the protective effect of NO. These data indicate the involvement of the GC-cGMP-PKG signaling pathway in NO-dependent cell protection against PDT-induced apoptosis. In contrast, Gupta et al. (1998) reported the involvement of NO in the apoptosis of A431 cells photosensitized with phthalocyanine Pc 4. Within the first 5 min after treatment, PDT increased the expression of the constitutive, but not the inducible form, of nitric oxide synthase, and NO production increased during the 15 min post-treatment. This effect was not observed in apoptosis-resistant fibrosarcoma RIF-1 cells. DiVenosa et al. (2005), however, did not find any cross-resistance between NO production and ALA-PDT in murine mammary adenocarcinoma cell lines producing or not producing NO, and resistant or not resistant to NO cytotoxicity. Therefore, the role of NO in cell resistance to photodynamic injury is cell-specific and may be realized through different signaling pathways.
Conclusion
Photodynamic therapy is an excellent example of the "magic bullet" concept suggested about a century ago by Paul Erlich, an outstanding biologist and physician, and founder of chemotherapy. Photosensitizers are selectively accumulated in the pathological tissue, which is then destroyed by light. The studies of the last 10-15 years have demonstrated that the "flight" of this "magic bullet" may be guided. Cells contain a lot of regulatory systems, signaling pathways that control apoptotic and even necrotic death.
The present data show that practically all signaling pathways are involved in responses of different cells to photodynamic injury. Some of them are aimed at cell protection, whereas others lead to cell death. Because of the complexity, differences between cells, cross-interactions, dependence on the environmental conditions and cellular functional state, it is not easy to recognize the real sequence of events leading to death of the given cell. This is why the current data on the role of signal transduction processes in various cell functions are so fragmentary. Future investigation should decipher unresolved mechanisms mentioned in the present chapter, thoroughly study the role of new signaling pathways that are still unknown, and explore the role of the interaction of different signaling pathways. The processes being upstream and downstream from the studied signaling protein are still largely unknown. The disclosure of the set of transcription factors and executioner proteins specific for each cell, which determine the physiological state of this cell and its response to external impact, is among the unresolved and the most difficult problems. A similar situation is in the studies of thebiological action of other physical and chemical factors, including ultraviolet and ionizing radiation, oxidative damage, and the action of various chemical agents.
New sophisticated technologies are being developed that reveal the expression of hundreds and thousands of genes and proteins, instead of the more primitive methods that provided the investigation of only a few signaling proteins in the cell. The application of genomic, transcriptomic, and proteomic approaches have shown a new perspective in the study of the complex signaling system in living cells when responding to photodynamic and other treatments. These methods include two dimensional electrophoresis, mass spectrometry, antibody, oligonucleotide, and cDNA microarrays, real time PCR, etc. (Wild et al., 2005; Cekaite et al., 2007; Ruhdorfer et al., 2007; Bhuwaneswatri et al., 2008; Buytaert et al., 2008; Tsaytler et al., 2008). For example, oligonucleotide microarray analysis has demonstrated the upregulation of 826 genes and the dowregulation of 653 genes involved in various metabolic processes, such as, stress-induced death, autophagy, proliferation, inflammation and carcinogenesis in T24 bladder cancer cells photosensitized with hypericin (Buytaert et al., 2008).
Another important outcome of these studies is opening the possibility of controlling pharmacologically the fate of photosensitized cells. Modulation of different signaling pathways can either enhance damage, or protect cells. Both effects are of significance for medical applications: using pharmacological modulators of different signaling pathways, one can increase the damage of malignant cells, or protect the surrounding healthy cells, or change the mechanism of cell death. Photodynamic therapy in combination with signaling modulators will take a place in a variety of medical treatment instruments in the near future.
Table 1. The most frequent abbreviations
AC = Adenylate cyclase
AIF = Apoptosis inducing factor
ANT = Adenine nucleotide translocator
AP-1 = Activating protein-1
APAF-1 = Apoptotic protease activating factor 1
ASK1 = Apoptosis signal-regulating kinase-1
CaMKII = Calmodulin-dependent kinase II
ΔΨm = Mitochondrial transmembrane potential
ER = Endoplasmic reticulum
ERK = Extracellularly regulated kinase
HIF-1 = Hypoxia-inducible factor
IAP = Apoptosis inhibitor protein
IMM = Inner mitochondrial membrane
JNK/SAPK = c-Jun terminal kinase/stress-activated
protein kinase
MAPK = Mitogen-activated protein kinase
mROS = Mitochondrial reactive oxygen species
OMM = Outer mitochondrial membrane
PBR = Peripheral benzodiazepine receptors
PI 3-kinase = Phosphatidylinositol 3-kinase
PKA = Protein kinase A
PKB = Protein kinase B/Akt
PKC = Protein kinase C
PLA2 = Phospholipase A2
PTP = Mitochondrial permeability transition pore
RCGP = Receptors coupled to G-proteins
ROS = Reactive oxygen species
RTK = Receptor tyrosine kinase
STAT = Signal transducer and activator of transcription
References
Agarwal M.L., Larkin H.E., Zaidi S.I., Mukhtar H., Oleinick N.L. (1993) Phospholipase activation triggers apoptosis in photosensitized mouse lymphoma cells, Cancer Res. 53: 5897–5902.
Aggarwal BB, Sethi G, Nair A, Ichikawa H. (2006) Nuclear factor-κB: a holy grail in cancer prevention and therapy. Curr Sign Transduct Therapy 1: 25-52.
Agostinis P., Donella-Deana A., Cuveele J, Vandenbogaerde A., Sarno S., Merlevede W., De Witte P. (1996) A comparative analysis of the photosensitized inhibition of growth factor regulated protein kinases by hypericin-derivatives, Biochem. Biophys Res Communs 220: 613-617.
Ahmad N., Feyes D.K., Agarwal R., Mukhtar H. (1998) Photodynamic therapy results in induction of WAF1/CIP1/P21 leading to cell cycle arrest and apoptosis. Proc Natl Acad Sci USA. 95:6977– 6982.
Ahmad N., Gupta S., Mukhtar H. (1999) Involvement of retinoblastoma (Rb) and E2F transcription factors during photodynamic therapy of human epidermoid carcinoma cells A431, Oncogene 18:1891–1896.
Ahmad N., Gupta S., Feyes D.K., Mukhtar H. (2000) Involvement of Fas (APO-1/CD-95) during photodynamic-therapy-mediated apoptosis in human epidermoid carcinoma A431cells, J. Invest. Dermatol. 115: 1041–1046.
Almeida R. D., Manadas B. J., Carvalho A. P., Duarte C. B. (2004) Intracellular signaling mechanisms in photodynamic therapy. Biochim. Biophys. Acta 1704: 59– 86.
Anis Y. (2006) Involvement of Ca2+ in the apoptotic process – “Friends or Foes”. Pathways; No 2: 1-7.
Assefa Z, Vantieghem A, Declercq W, Vandenabeele P, Vandenheede JR, Merlevede W, de Witte P, Agostinis P. (1999) The activation of the c-Jun N-terminal kinase and p38 mitogen-activated protein kinase signaling pathways protects HeLa cells from apoptosis following photodynamic therapy with hypericin, J Biol Chem. 274: 8788– 8796.
Barberi-Heyob M, Vedrine PO, Merlin JL, Millon R, Abecassis J, Poupon MF, Guillemin F. (2004) Wild-type p53 gene transfer into mutated p53 HT29 cells improves sensitivity to photodynamic therapy via induction of apoptosis. Int J Oncol. 24:951-958.
Belzacq A.S., Jacotot E., Vieira H.L., Mistro D., Granville D.J., Xie Z., Reed J.C., Kroemer G., Brenner C. (2001) Apoptosis induction by the photosensitizer verteporfin: identifcation of mitochondrial adenine nucleotide translocator as a critical target. Cancer Res. 61: 1260–1264.
Ben-Hur E, Dubbelman TM. (1993) Cytoplasmic free calcium changes as trigger mechanism in the response of cells to photosensitization. Photochem Photobiol 58: 890-894.
Bhuvaneswari R, Gan YY, Lucky SS, Chin WW, Ali SM, Soo KC, Olivo M. (2008) Molecular profiling of angiogenesis in hypericin mediated photodynamic therapy. Mol Cancer. 13; 56, 1-14.
Bragin D.E., Kolosov M.S., Uzdensky A.B. (2003) Photodynamic inactivation of isolated crayfish neuron requires protein kinase C, PI 3-kinase and Ca2+. J. Photochem. Photobiol. B: Biol. 70: 99-105.
Bras M., Queenan B., Susin S.A. (2005) Programmed cell death via mitochondria: different models of dying. Biochemistry (Moscow), 70, 231-239.
Buytaert EP, Callewaert G, Hendrickx N, Scorrano L, et al. (2006) Role of endoplasmic reticulum depletion and multidomain proapoptotic Bax and Bak proteins in shaping cell death after hypericin-mediated photodynamic therapy. FASEB J; 20:756-768.
Buytaert E, Matroule JY, Durinck S, Close P, Kocanova S, Vandenheede JR, de Witte PA, Piette J, Agostinis P. (2008) Molecular effectors and modulators of hypericin-mediated cell death in bladder cancer cells. Oncogene.27:1916-1929.
Castano AP, Demidova TN, Hamblin MR. (2005) Mechanisms in photodynamic therapy: part two – cellular signaling, cell metabolism and modes of cell death. Photodiagn Photodyn Ther. 2: 1-23.
Cekaite L, Peng Q, Reiner A, Shahzidi S, Tveito S, Furre IE, Hovig E. (2007) Mapping of oxidative stress responses of human tumor cells following photodynamic therapy using hexaminolevulinate. BMC Genomics. 8:273,1-20.
Chan WH, Yu JS, Yang SD. (2000) Apoptotic signaling cascade in photosensitized human epidermoid carcinoma A431 cells: invovement of singlet oxygen, c-Jun N-terminal kinase, caspase-3 and p21-activated kinase 2. Biochem. J, 351: 221-232.
Finkel T. (1998) Oxygen radicals and signaling. Curr. Opin. Cell Biol. 10, 248-253. Fisher A.M., Ferrario A., Rucker N., Zhang S., Gomer C.J. (1999) Photodynamic therapy sensitivity is not altered in human tumor cells after abrogation of p53 function, Cancer Res. 59: 331– 335.
Girotti A.W., Kriska T. (2004) Role of lipid hydroperoxides in photooxidative stress signaling. Antioxidants Redox Signal. 6: 301-310.
Glinski J.A., David E., Warren T.C., Hasen G., Leonard S.F., Pitner P., Pav S., Arvigo R., Balick M.J., Panti E., Grob P.M. (1995) Inactivation of cell surface receptors by pheophorbide a, a green pigment isolated from Psichotria acuminata. Photochem. Photobiol. 62: 144-150.
Gomes E.R., Almeida R.D., Carvalho A.P., Duarte C.B. (2002) Nitric oxide modulates tumor cell death induced by photodynamic therapy through a cGMP-dependent mechanism, Photochem. Photobiol. 76: 423–430.
Granville D.J., Jiang H, An M.T., Levy J.G., McManus B.M., Hunt D.W. (1999a) Bcl-2 overexpression blocks caspase activation and downstream apoptotic events instigated by photodynamic therapy. Br. J. Cancer 79: 95-100.
Granville D.J., Carthy C.M., Jiang H., Levy J.G., McManus B.M., Matroule J.Y., Piette J., Hunt D.W. (2000) Nuclear factor-kappa B activation by the photochemotherapeutic agent verteporfin, Blood 95: 256–262.
Granville DJ, Ruehlmann DO, Choy JC, Cassidy BA, Hunt DW, van Breemen C, McManus BM. (2001) Bcl-2 increases emptying of endoplasmic reticulum Ca2+ stores during photodynamic therapy-induced apoptosis. Cell Calcium. 30:343-350.
Grebenova D., Kuzelova K., Smetana K., Pluskalova M., Cajthamlova H., Marinov I., Fuchs O., Soucek J., Jarolim P., Hrkal Z. (2003) Mitochondrial and endoplasmic reticulum stress-induced apoptotic pathways are activated by 5-aminolevulinic acid-based photodynamic therapy in HL60 leukemia cells, J Photochem Photobiol. B. Biol. 69: 71–85.
Gupta S, Ahmad N, Mukhtar H. (1998) Involvement of nitric oxide during phthalocyanine (Pc4) photodynamic therapy-mediated apoptosis. Cancer Res 58: 1785-1788.
Gupta S, Dwarakanath BS, Muralidhar K, Jain V. (2003) Role of apoptosis in photodynamic sensitivity of human tumour cell lines. Indian J Exp Biol. 41:33-40. Heinzelmann-Schwarz V, Fedier A, Hornung R, Walt H, Haller U, Fink D. (2003) Role of p53 and ATM in photodynamic therapy-induced apoptosis. Lasers Surg Med. 33:182-189.
Hendrickx N., Volanti C., Moens U., Seternes O.M., De Witte P., Vandenheede J.R., Piette J., Agostinis P. (2003) Up-Regulation of cyclooxygenase-2 and apoptosis resistance by p38 mapk in hypericin mediated photodynamic therapy of human cancer cells, J. Biol. Chem. 278: 52231– 52239.
Hensley K., Robinson K.A., Gabbia P., Salsman S., Floyd R.A.(2000) Reactive oxygen species, cell signaling and cell injury. Free Rad. Biol. Med. 28: 1456-1462.
Hsien Y.-J., Wu C.-C., Chang C.-J., Yu J.-S. (2002) Subcellular localization of photofrin determines the death phenotype of human epidermoid carcinoma a431 cells triggered by photodynamic therapy: when plasma membranes are the main targets. J. Cell Physiol. 194: 363-375.
Inanami O., Yoshito A., Takahashi K., Hiraoka W., Kuwabara M. (1999) Effects of bapta-am and forskolin on apoptosis and cytochrome c release in photosensitized chinese hamster v79 cells, Photochem. Photobiol. 70: 650–655.
Ji Z, Yang G, Shahzidi S, Tkacz-Stachowska K, Suo Z, Nesland J, Peng Q. (2006) Induction of hypoxia-inducible factor-1alpha overexpression by cobalt chloride enhances cellular resistance to photodynamic therapy. Cancer Lett. 244:182-189.
Johnson G.L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, Jnk, and p38 protein kinases. Science 298: 1911-1913.
Kessel D., Antonovich M., Smith K.M. (2001) The role of the peripheral benzodiazepine receptor in the apoptotic response to photodynamic therapy. Photochem. Photobiol. 74: 346-349.
Kim H.R., Luo Y., Li G., Kessel D. (1999) Enhanced apoptotic response to photodynamic therapy after Bcl-2 transfection, Cancer Res. 59: 3429–3432. Klein SD, Walt H, Richter C. (1997) Photosensitization of isolated rat liver mitochondria by tetra(m-hydroxyphenyl)chlorin. Arch Biochem Biophys. 348 : 313-319.
Koukourakis MI, Giatromanolaki A, Skarlatos J, Corti L, Blandamura S, Piazza M, Gatter KC, Harris Al. (2001) Hypoxia inducible factor (hif-1α and hif-2α) expression in early esophageal cancer and response to photodynamic therapy and radiotherapy. Cancer res.; 61: 1830-1832.
Lee HB, Ho AS, Teo SH. (2005) P53 status does not affect photodynamic cell killing induced by hypericin. Cancer Chemother Pharmacol. 7:1-8.
Liu W, Oseroff Ar, Baumann H. (2004) Photodynamic therapy causes cross-linking of signal transducer and activator of transcription proteins and attenuation of interleukin-6 cytokine responsiveness in epithelial cells. Cancer Res.; 64: 6579-6587.
Luna M.C., Wong S., Gomer C.J. (1994) Photodynamic therapy mediated induction of early response genes, Cancer Res. 54:1374–1380.
Matroule JY, Piette J (2000) Photosensitization and redox signaling. Antioxidants Redox Signal 2: 301-315.
Matroule JY, Carthy CM, Granville DJ, Jolois O, Hunt DW, Piette J. 2001 Mechanism of colon cancer cell apoptosis mediated by pyropheophorbide-a methylester photosensitization. Oncogene. 20: 4070-4084.
Moan J., Juzeniene A. (2008) The riole of oxygen in photodynamic therapy. In: Advances in photodynamic therapy. M.R. Hamblin and P. Mroz (Eds). Artech house: Boston-London, 135-149.
Moreno G., Poussin K., Ricchelli F., Salet C. (2001) The effect of singlet oxygen produced by photodynamic action on the mitochondrial permeability transition differ in accordance with the localization of the sensitizer. Arvh. Biochem. Biophys. 386: 243-250.
Niziolek M., Korytowski W., Girotti A.W. (2003) Chain-breaking antioxidant and cytoprotective action of nitric oxide on photodynamically stressed tumor cells, Photochem. Photobiol. 78: 262– 270.
Nowis D., Makowski M., Stoklosa T., Legat M., Issat T., Golab J. (2005) Direct tumor damage mechanisms of photodynamic therapy. Acta Biochim. Pol. 52: 339-352.
Ogata M., Inanami O., Nakajima M., Nakajima T., Hiraoka W., Kuwabara M. (2003) Ca2+-dependent and caspase-3-independent apoptosis caused by damage in golgi apparatus due to 2,4,5,7-tetrabromorhodamine 123 bromide-induced photodynamic effects, Photochem. Photobiol. 78: 241–247.
Oleinick N.L., Morris R.L., Belichenko I. (2002) The role of apoptosis in response to photodynamic therapy: what, where, why, and how. Photochem. Photobiol. Sci. 1: 1-21.
Penning L.C., VanSteveninck J., Dubbelman T.M. (1993a) HPD-induced photodynamic changes in intracellular cyclic AMP levels in human bladder transitional carcinoma cells, clone T24, Biochem. Biophys. Res. Commun. 194: 1084– 1089.
Penning LC, Keirse MJ, VanSteveninck J, Dubbelman TM. (1993b) Ca2+-mediated prostaglandin E2 induction reduces haematoporphyrin-derivative-induced cytotoxicity of T24 human bladder transitional carcinoma cells in vitro, Biochem. J. 292: 237–240.
Pollack I.F., Kawecki S. (1997) The effect of calphostin C, a potent photodependent protein kinase inhibitor, on the proliferation of glioma cells in vitro. J. Neuro-Oncol. 31: 255-266.
Rash MH, Tijssen K, Lagerberg JW, Corver WE, VanSteveninck J, Dubbelman TM. (1997) The role of protein kinase C activity in the killing of Chinese hamster ovary cells by ionizing radiation and photodynamic treatment, Photochem. Photobiol. 66: 209– 213.
Reiners J.J., Caruso J.A., Mathieu P., Chelladurai B., Yin X.-M., Kessel D. (2002) Release of cytochrome C and activation of pro-caspase-9 following lysosomal photodamage involves bid cleavage. Cell Death Diff. 9: 934-944.
Ricchelli F, Barbato P, Milani M, Gobbo S, Salet C, Moreno G. 1999 Photodynamic action of porphyrin on Ca2+ influx in endoplasmic reticulum: a comparison with mitochondria. Biochem J. 338:221-227.
Rodriguez-Mora OG, Lahair MM, Evans MG, Kovacs CJ, Allison RR, Sibata CH, White KS, McCubrey JA, Franklin RA. (2006) Inhibition of CaM-kinases augments cell death in response to oxygen radicals and oxygen radical inducing cancer therapies in MCF-7 human breast cancer cells. Cancer Biol Ther., 5:1022-1030.
Ruhdorfer S, Sanovic R, Sander V, Krammer B, Verwanger T. (2007) Gene expression profiling of the human carcinoma cell line A-431 after 5-aminolevulinic acid-based photodynamic treatment. Int J Oncol. 30:1253-1262.
Ryter S.W., Gomer C.J. (1993) Nuclear factor kappa B binding activity in mouse L1210 cells following photofrin II-mediated photosensitization, Photochem. Photobiol. 58: 753–756.
Salet C., Moreno G., Ricchelli F. (1997) Effect of Photofrin photodynamic action on mitochondrial respiration and superoxide radical generation. Free Rad. Res.26: 201-208.
Schieke SM, VonMontfort C, Buchczyk DP, Timmer A, Ghether-Beck S, Krutmann J, Holbrook NJ, Klotz LO. (2004) Singlet oxygen-induced attenuation of growth factor signaling: Possible role of ceramides. Free Rad Res; 38: 729-737.
Shen XY, Zacal N, Singh G, Rainbow AJ. (2005) Alterations in mitochondrial and apoptosis-regulating gene expression in photodynamic therapy-resistant variants of HT29 colon carcinoma cells. Photochem Photobiol. 81:306-313.
Skulachev V.P. (1999) Mitochondrial physiology and pathology; concepts of programmed death of organelles, cells, and organisms. Mol. Aspect.Med. 20: 139-184.
Srivastava M., Ahmad N., Gupta S., Mukhtar H. (2001) Involvement of Bcl-2 and Bax in photodynamic therapy-mediated apoptosis. Antisense Bcl-2 oligonucleotide sensitizes RIF1 cells to photodynamic therapy apoptosis, J. Biol. Chem. 276: 15481–15488.
Takahashi H, Itoh Y, Miyauchi Y, Nakajima S, Sakata I, Ishida-Yamamoto A, Iizuka H. (2003) Activation of two caspase cascades, caspase 8/3/6 and caspase 9/3/6, during photodynamic therapy using a novel photosensitizer, ATX-S10(Na), in normal human keratinocytes. Arch Dermatol Res. 295:242-248.
Tong Z., Singh G., Rainbow A.J. (2000) The role of the p53 tumor suppressor in the response of human cells to photofrin-mediated photodynamic therapy, Photochem. Photobiol. 71: 201–210.
Tong Z., Singh G., Rainbow A.J. (2002) Sustained activation of the extracellular signal regulated kinase pathway protects cells from Photofrin-mediated photodynamic therapy. Cancer Res. 62: 5528– 5535.
Tsaytler PA, C O'Flaherty M, Sakharov DV, Krijgsveld J, Egmond MR. (2008) Immediate Protein Targets of Photodynamic Treatment in Carcinoma Cells. J Proteome Res. Jul 25. [Epub ahead of print]
Turpaev K.T. (2002) Reactive oxygen species and regulation of gene expression. Biochemistry (Mosc). 67:281-292.
Usuda J., Chiu S.M., Azizuddin K., Xue L.Y., Lam M., Nieminen A.L., Oleinick N.L. (2002) Promotion of photodynamic therapy-induced apoptosis by the mitochondrial protein Smac/DIABLO: dependence on Bax, Photochem. Photobiol. 76: 217–223.
Uzdensky A. B., Zhavoronkova A. A., Dergacheva O. Y. (2000) Firing inhibition processes in the response dynamics of isolated crayfish nerve cell to the photodynamic effect of sulphonated aluminum phthalocyanine: participation of free radicals and Ca2+. Lasers Med. Sci. 15: 123-130.
Uzdensky A., Bragin D., Kolosov M., Dergacheva O., Fedorenko G., Zhavoronkova A. (2002) Photodynamic inactivation of isolated crayfish mechanoreceptor neuron. Photochem. Photobiol. 76: 431-437.
Uzdensky A., Kolosov M., Bragin D., Dergacheva O., Vanzha O., Oparina L. (2005) Involvement of adenylate cyclase and tyrosine kinase signaling pathways in response of crayfish stretch receptor neuron and satellite glia cell to photodynamic treatment. Glia 49: 339-348.
Uzdensky A., Lobanov A., Bibov M., Petin Y. (2007) Involvement of Ca2+- and cyclic adenosine monophosphate-mediated signaling pathways in photodynamic injury of isolated crayfish neuron and satellite glial cells. J. Neurosci. Res., 85: 860-870.
Uzdensky A.B. (2008) Signal Transduction and Photodynamic Therapy. - Current Signal Transduction Therapy. 3: 55-74.
Vantieghem A., Xu Y., Declercq W., Vandenabeele P., Denecker G., Vandenheede J.R., Merlevede W., de Witte P.A., Agostinis P. (2001) Different pathways mediate cytochrome c release after photodynamic therapy with hypericin, Photochem. Photobiol. 74: 133–142.
Di Venosa G, Casas A, Fukuda H, Perotti C, Batlle A. (2005) No cross-resistance between ALA-mediated photodynamic therapy and nitric oxide. Nitric Oxide. 13:155-162.
Volanti C, Matroule JY, Piette J. (2002) Involvement of oxidative stress in NFκB activation in endothelial cells treated by photodynamic therapy. Photochem Photobiol 75: 36-45.
Volanti C, Hendrickx N, Van Lint J, Matroule JY, Agostinis P, Piette J. (2005) Distinct transduction mechanisms of cyclooxygenase 2 gene activation in tumour cells after photodynamic therapy. Oncogene. 24: 2981-2991.
Weller M., Trepel M., Grimmel C., Schabet M., Bremen D., Krajewski S., Reed J.C. (1997) Hypericin-induced apoptosis of human malignant glioma cells is light-dependent, independent of bcl-2 expression, and does not require wild-type p53. Neurol Res, 19, 459-470.
Wild PJ, Krieg RC, Seidl J, Stoehr R, Reher K, Hofmann C, Louhelainen J, Rosenthal A, Hartmann A, Pilarsky C, Bosserhoff AK, Knuechel R. (2005) RNA expression profiling of normal and tumor cells following photodynamic therapy with 5-aminolevulinic acid-induced protoporphyrin IX in vitro. Mol Cancer Ther. 4:516-528.
Wong T.W., Tracy E., Oseroff A.R., Baumann H. (2003) Photodynamic therapy mediates immediate loss of cellular responsiveness to cytokines and growth factors, Cancer Res. 63: 3812– 3818.
Xue L-Y., Qiu Y., He J., Kung H-J., Oleinick N.L. (1999a) Etk/Bmx, a PH-domain containing tyrosine kinase, protects prostate cancer cells from apoptosis induced by photodynamic therapy or thapsigargin, Oncogene 18: 3391-3398.
Xue L., He J., Oleinick N.L. (1999b) Promotion of photodynamic therapy induced apoptosis by stress kinases, Cell Death Differ. 6: 855–864.
Yu J, Zhang L. (2005) The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Communs 331: 851–858.
Zhuang S., Lynch M.C., Kochevar I.E. (1998) Activation of protein kinase C is required for protection of cells against apoptosis induced by singlet oxygen, FEBS Lett. 437: 158-162
Zhuang S., Demirs J.T., Kochevar I.E. (2000) p38 Mitogen-activated protein kinase mediates Bid cleavage, mitochondrial dysfunction, and caspase-3 activation during apoptosis induced by singlet oxygen but not by hydrogen peroxide, J. Biol. Chem. 275: 25939–25948.
Zhuang S., Kochevar I.E. (2003) Singlet oxygen-induced activation of Akt/protein kinase B is independent of growth factor receptors, Photochem. Photobiol. 78: 361– 371.
8/19/08