BYSTANDER EFFECT INDUCED BY UV RADIATION
Maria Widel
Biotechnology Center, Institute of Automatic Control
Silesian University of Technology, Akademicka 16 Street
44-100 Gliwice, Poland
maria.widel@polsl.pl
The radiation-induced bystander effect is defined as the induction of biological changes in non-irradiated cells by signals transmitted from irradiated neighboring cells. Although the phenomenon was originally attributed to ionizing radiation (e.g., Prise et al., 2003; Rzeszowska-Wolny et al., 2009), it also occurs in the case of other stressors, including ultraviolet radiation (UVR) (Widel, 2012), chemotherapeutic drugs (Alexandre et al., 2007), and photodynamic treatment (Dahle et al., 2000; Chakraborty et al., 2009).
UVR comprises three different bands; long-wave UVA (320-400 nm), middle-wave UVB (290-320 nm), and short-wave UVC (200-290 nm) (Hockberger, 2002), and increasing data reveal that all three bands are able to induce the bystander effect. About 95% of solar radiation, which is the main source of UVR in environment is UVA and about 5% is UVB. UVC is almost entirely absorbed in the upper part of the stratosphere (Hockberger, 2002), however, it comes also from artificial sources. The short-wave radiations (UVB under 300 nm and UVC) are particularly dangerous for cells because their bands overlap with the absorption spectra of DNA, RNA and proteins, and they can damage DNA by forming cyclobutane pyrimidine dimers and 6-4 photoproducts, which can lead indirectly to DNA strand breaks (Wang et al., 1986; Sliman et al., 2000; Rastogi et al., 2010), and to mutations and carcinogenesis (Rünger, 2007). UVR is considered to be responsible for induction and promotion of basal and squamous cell skin cancer (de Grujil et al. 1993), and is also an important etiological factor for malignant melanoma (Setlow et al., 1993).
Because the bystander effect is usually manifested as damaging events, we can expect that it also has its share in carcinogenesis. Data about the possible role of bystander effect(s) in UV carcinogenesis come exclusively from in vitro studies, however, up-to-date data on UV-induced bystander effects in vivo are not available. This review summarizes the existing studies on bystander effects induced by three bands of UV radiation with emphasis placed on cellular effects, mechanisms of action and similarities and differences in results observed.
Cellular Events of the Bystander Effect Induced by UVA, UVB and UVC Radiation
The bystander effect observed in cells resembles the effects observed in directly exposed cells (Figure 1). These responses are observed in cells that are in the vicinity of the irradiated cells (horizontal transmission of bystander signals), or in subsequent generations of irradiated cells (vertical transmission) (Mothersill and Seymour, 2012). The vertical conveyance of signals from the irradiated cells to their progenies is defined as genomic instability, and delayed cell death (Kadim et al. 1995; Dahle and Kvam, 2003).
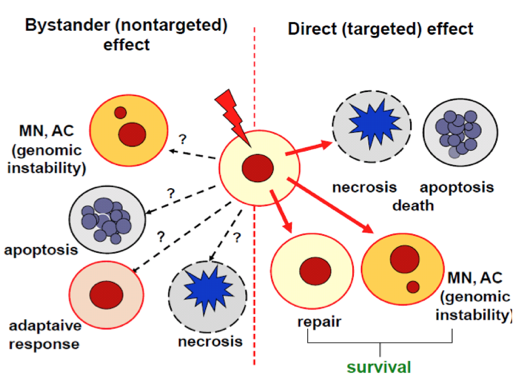
Figure 1. Illustration of the cellular effects caused by the direct exposure of cells to radiation (ionizing, UV), and the bystander effects resulting from the operation of molecular signals secreted by irradiated cells. Question marks represent not fully recognized molecular signals which induce changes in non-irradiated cells; MN, micronuclei; AC, chromosomal aberrations.
The experimental models frequently used to study the bystander effect are the medium transfer system and the co-incubation transwell system (Figure 2).
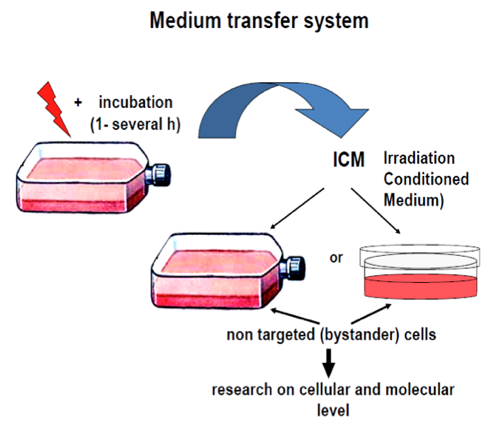
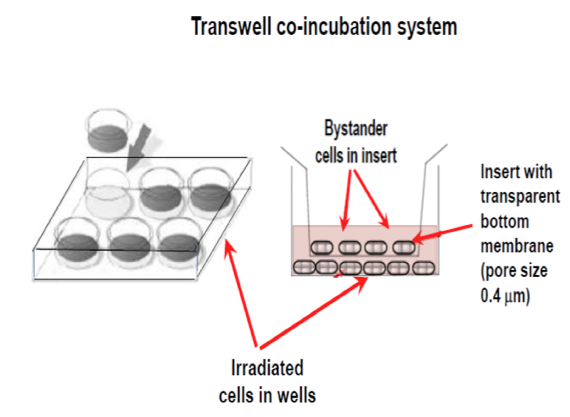
Figure 2. The two main experimental systems used for studying the bystander effect; the Medium Transfer System and the Transwell Co-incubation System.
In the first case, the medium harvested from irradiated cells, containing any signaling molecules is collected at various times after irradiation, filtered and transferred to a non-irradiated cell culture. The second system allows for the free flow of signals between irradiated and bystander cells through a semi-permeable, transparent membrane constituting the bottom of the insert, but not allowing for the direct contact of the cells. This system is more relevant to situation in vivo, since both type of cells may mutually exchange signals for a longer period of time. Other experimental systems are also adapted to examine the bystander effect upon UV exposure, for example, half-shielding of the culture vessel (Dickey et al., 2008).
The most commonly observed cellular events of the bystander effect are: reduction in survival, induction of apoptosis and genomic instability, including early and delayed mutations, chromosomal aberrations, micronuclei formation and delayed cell death (Widel, 2012).
Bystander Effect-Mediated Genomic Instability
UVA and UVB radiations both induce immediate and delayed mutations, however, with different efficiency. Delayed mutations are especially indicative of GI. Human keratinocytes, HaCaT line, whose precursory generations were exposed to 100 kJ/m2 UVA showed a continuing reduction in plating efficiency, reduction of clonogenic cell survival, increase of micronuclei formation and delayed mutation of the hypoxanthine-phosphoribosyl-transferase (hprt) gene observed up to 21 days after the initial exposure, corresponding to approximately the 23rd division (Phillipson et al., 2002).
A reduction in survival of clonogenic cells and the mutation of the hprt locus has been observed in bystander V79 Chinese hamster fibroblasts after exposure to UVA (265 kJ/m2) and UVB (9.1 kJ/m2), the doses killing 80% of the cells (Dahle and Kvam, 2003). UVA radiation produced the lower level of immediate mutations in the hprt locus in V79 clones derived from single cells, which survived radiation, but higher level of delayed mutations than UVB. On the other hand, UVB was very effective in the induction of immediate mutations, even more effective than X-ray radiation (1 Gy). Furthermore, UVB induced about 5% of centrosome aberrations, whereas UVA did not, but caused elevation in chromosome number (Dahle and Kvam, 2003). These results also indicate that mechanisms leading to genomic instability for both of these UV radiations differ substantially. Later studies of Dahle et al. (2005a) using a modified protocol on the same cell line showed that UVB was ~5-fold more effective in the induction of delayed mutations than was UVA, and this effect was reduced by antioxidant treatment (Dahle et al., 2005b).
However, the short term and delayed mutagenic events in a critical genes, as well as numerical chromosome aberrations may all be the first steps in carcinogenesis, and may pose a potential risk to human health. Persistent genomic instability manifested as a prolonged reduction in plating efficiency and lethal mutation was also reported by O’Reilly and Mothersill (1997) after the UVA and UVB exposure of human skin cells, but was absent in fish epithelial cells (EPC line). In contrast, EPC cells appeared to be the more sensitive to direct UVA and UVB radiation than human keratinocytes when clonogenic survival was used as the endpoint.
Thus, it appears that the induction of genomic instability is dependent on the UV spectrum, the dose used and cell types, and may vary depending on the assessment criterion.
Cell Survival, Apoptosis, Senescence and DNA Damage in Bystander Cells Induced by UVA, UVB and UVC Radiation
The study of Whiteside and McMillian (2009) performed on HaCaT human keratinocytes and MRC5 fibroblasts, aimed at the comparison of the bystander effect after UVA and UVB, did not show a reduction of clonogenic bystander cells after 400 J/m2 UVB radiation. However, the relatively low dose of UVA (100 kJ/m2, equivalent to 30 min of summer midday sun exposure) significantly reduced the clonogenicity of bystander cells co-incubated with irradiated cells in the medium-permeable inserts. This effect was induced within and between the two cell lines.
It should be emphasized, that following exposure to UVA, the bystander effect occurs not only within several hours after exposure, but it also shows up even days post exposure (Whiteside et al., 2011). Using the co-incubation system, the authors found that HaCaT cells exposed to 100 kJ/m2 UVA induced the bystander effect in the unirradiated recipient cells when co-incubation was started 3 days post irradiation. This effect appeared as a reduction in plating efficiency and clonogenic survival. This means that bystander signals generated by a single UVA dose, corresponding to the dose in the natural environment at a latitude of 48 degrees north, are continuously released by the irradiated cells, thus they may amplify the detrimental effects of UVA exposure.
Studies by Dickey et al. (2009) using the half-shielded protocol showed that the bystander effect was induced after exposure to UVA, both in normal NBFs fibroblasts, primary prostate epithelial cells (PrECs), and HeLa cancer cells. All three cell types experienced bystander effects in response to UVA radiation. However, there were essential differences in the timing and features of the bystander response, e.g., the maximal increase in double strand breaks (DSBs) visualized as γH2AX/53BP1 foci varied from 3 days post-exposure for fibroblasts to 3 h post-exposure for PrECs and HeLa cells. The half-shielded bystander culture of PrEC cells exhibited about a 7-fold increase in the numbers of γH2AX/53BP1 foci 3 h post UVA irradiation, whereas HeLa cells showed a ~3-fold increase of γH2AX/53BP1 foci 1 day post exposure that further increased at 5 days, mainly due to apoptosis. In contrast to HeLa cells, bystander cultures of prostate cells contained only minor level of apoptosis. However, they did contain many senescent cells. Bystander NBFs cells exhibited a ~3-fold increase of γH2AX levels over control cultures after 3 days. These variations in responses may indicate the difference between normal and cancer cells in receiving bystander stress signaling (Dickey at al., 2009).
In our study on NHDF fibroblasts exposed to UVA (20 kJ/m2), UVB (10 kJ/m2) and UVC (200 J/m2), the diminution of viability of bystander cells, estimated in MTS assay, was roughly comparable for UVA and UVC (~45% of control survival) and slightly lower for UVB (~60% of control survival) (Widel et al., 2014). However, UVA and UVB were more effective in apoptosis induction in irradiated and bystander cells measured by Annexin V expression, whereas UVC induced apoptosis mainly in irradiated cells. In contrast, UVC efficiently induced premature senescence not related to telomere shortening (Suzuki et al., 2008) in both directly exposed and bystander cells. UVA generated senescence only in directly exposed cells, but UVB in bystanders. These differences suggest that signals secreted by cells in response to different UV bands differ, although their nature and interaction needs to be elucidated.
Apoptosis was also observed in bystander human HaCaT keratinocytes in the medium transfer system after exposure to mix light, UVA (3.5 kJ/m2) + UVB (300 J/m2) (Banerjee et al., 2005). The apoptosis was visualized by reduction of viability, DNA laddering, and pro-apoptotic genes Bax and Fas expression. The dose of UVA was, however, very low and could be neglected, thus authors believed that apoptosis was induced in bystander cells mainly by UVB-generated secreted factors. The nature of these factors was however not defined, although, the nerve growth factor was suggested as one of the possible signaling molecules, which has a different affinity to the cell surface specific receptors, p75 and trkA, associated with survival and death (Banerjee et al., 2005). Apoptosis in UV exposed cells is a complex event, and can go via various ways. Signaling pathways involved in UVB-induced apoptosis may be joined with DNA damage, direct activation of death receptors and formation of reactive oxygen species (Kulms et al., 2002). However, in experiments of Banerjee et al. (2005), the generation of ROS in bystander cells was not found, thus, an induction of apoptosis in bystander cells could be a consequence of an operation of other, not yet discovered signaling molecules. Interestingly, the large dilution of conditioned medium did not induce apoptosis, but stimulated proliferation, indicating the complexity of the bystander signaling and response.
The bystander effect, in the case of UVC, has been less investigated. In addition to our research, outlined above, we investigated the bystander effect after the exposure of K562 leukemia cells to UVC (100-500 J/m2), and found increased levels of micronuclei in bystander cells, even exceeding those observed in cells directly exposed (Widel, 2012). Furthermore, we noticed an increase in DNA damage at a much lower dose (20 J/m2), as measured by the alkaline comet assay in bystander human malignant melanoma Me45 cells. Interestingly, no increase of DNA damage was noticed in NHDF fibroblasts in the comet assay, indicating a lower susceptibility of these normal cells to bystander signaling. Morever, the DSBs were also induced in bystander HCT116 colon cancer cells co-incubated with UVC-exposed cells, as judged by the expression of phosphorylated histone H2AX (γH2AX foci) (Widel, 2012).
DSBs visualized by γH2AX and 53BP1 focal formation (γH2AX/53BPI foci) was also found by Dickey et al. (2008) in bystanders of UVC-exposed normal human fibroblasts, HeLa cervical adenocarcinoma and primary prostate epithelial cells. Bystander fibroblasts showed increasing numbers and intensity of γH2AX foci after co-incubation with 20 J/m2 UVC irradiated cells, although there was no detectable increase in γH2AX focal numbers in the directly exposed cells. These results suggest that damage other than DSBs generated by UVC induce bystander signals that cause DNA DSBs in neighboring unirradiated cells. This DNA damage generated in unexposed cells was associated with an increase in apoptosis.
Recently, Wu et al. (2014) showed that medium collected from UVC-exposed (90 mJ/cm2) V79 fibroblasts reduced viability and induced apoptosis and chromosomal aberrations (mostly breaks) in bystander cells. Interestingly, the medium collected at 4 and 24 h post irradiation showed greater effects than when collected after 8 and 12 hours, suggesting that secretion of molecular signals may be also cell cycle dependent.
Putative Mechanism for the Bystander Effect; Reactive Oxygen Species are the Main Inducers of the Phenomenon
The three bands of UVR act in fundamentally different ways. UVA acts particularly in the presence of oxygen via the generation of reactive oxygen species (ROS), such as singlet oxygen and hydroxyl radicals and nitrogen species, like nitric oxide (Arimoto-Kobayashi et al., 2002; Phillipson et al., 2002; McMillan et al., 2008), which can induce oxidative damage to DNA, proteins, and lipids. UVB and UVC photons are absorbed by DNA and induce specific DNA damage: the cyclobutane pirimidine dimers and 6-4 photoproducts which lead to transition mutations C→T or CC→TT (Rünger, 2007). Accumulation of this type of DNA damage and their transmission to daughter cells may lead to neoplastic transformation.
However, UVB can also generate ROS leading to oxidative stress. Oxidative species induced by UVB appear particularly in the form of hydrogen peroxide and lipid peroxidation (Masaki et al., 1995; Morliere et al., 1995; Dahle et al. 2005a, 2005b), although hydrogen peroxide can also be generated by UVA (Phillipson et al., 2002). Malondialdehyde (MDA) and 4-hydroxy-nonenal (4HNE), the main lipid peroxidation by-products, are small molecules which are able to diffuse within and between cells, and can create adducts with proteins, DNA and phospholipids in bystander cells. DNA adducts are potentially mutagenic and carcinogenic agents (Marnett, 2000; Zhong et al, 2001). Furthermore, the end-products of lipid peroxidation also have properties of secondary messengers, which can activate both the signal cascade leading to the repair of DNA damage and to stabilizing them, or to apoptosis (Esterbauer et al., 1991).
Oxidative stress is an important mediator of bystander effects induced by ionizing radiation (Harada et al., 2008; Widel et al. 2012), therefore it can be expected that oxidative stress induced by UVR will also promote the bystander effect in cells. A number of investigators have presented data indicative of this up-regulation of cellular oxidative status as a driver of the bystander effect after UV radiation, since ROS scavengers reduced the bystander effect-mediated genomic instability and ameliorated its effect on survival reduction.
Phillipson et al. (2002) observed that pretreatment of keratinocytes with catalase (CAT) before exposure to UVA reduced genomic instability, which was observed as micronuclei formation, delayed cell death, and increased mutagenicity. The reduction of these effects by catalase suggest that hydrogen peroxide, and probably its downstream products (e.g., lipid peroxidation end-products) are mediators of this phenomenon.
Studies of Dahle et al. (2005b) have revealed that genomic instability, especially delayed hprt mutation induced by both the UVA and UVB, was considerably inhibited by antioxidants that eliminate hydrogen peroxide and superoxide radical anions. While, in the system applying UVB, strong inhibition of the bystander effect was achieved by the application of CAT, reduced glutathione (GSH) and superoxide dismutase (SOD); in the UVA system, only GSH was effective in the inhibition of delayed mutation. Neither SOD, nor CAT penetrate the cell membrane, thus it follows that in the case of UVB, bystander signals inducing delayed mutation are mediated via the medium or via gap junctional intercellular communication, and are also transmitted from mother to daughter cells. However, in the case of UVA, they had to be transmitted from mother to the daughter cells, or transferred by gap junctions.
Pretreatment of bystander HaCaT keratinocytes with an inhibitor of NADPH-oxidase, diphenyleneiodonium, led to the diminution of the bystander effect when measured as clonogenic cell survival (McMillan et al., 2008). The results point out a role of this enzyme, participating in ROS induction, in bystander signaling and place ROS as an initiator of bystander signals.
Our recent study performed on the human dermal fibroblasts (Widel et al., 2014), show that oxidative metabolism is deregulated after exposure to all three regions of UVR. After the irradiation of cells with UVA, the rapid increase in the frequency of apoptosis was paralleled by increases in ROS and superoxide radicals. Interestingly, UVC was more effective in the generation of ROS in bystanders than in irradiated cells, and produced a higher yield of superoxide in exposed and bystander cells in comparison with UVA and UVB radiation.
Moreover, several cytokines are considered to be potential mediators of the bystander effect. Yoshizumi et al. (2008) found that UVB at a dose of 400 mJ/cm2 induced the release of various cytokines; interleukins IL-1beta, IL-6, IL-8, interferon (IFN)-gamma, tumor necrosis factor (TNF)-alpha and others, which mediate the inflammatory response. Transforming growth factor beta (TGF-β), and nitric oxide (NO) were also proposed as mediators of the bystander response, since they elevated numbers of γH2AX/53BP1 foci in normal cell cultures comparably to the levels found in bystander cells, and this elevation was abrogated by nitric oxide synthase (iNOS) inhibitors, TGF-β blocking antibody and antioxidants (Dickey et al., 2008). Similarly, it was shown that the bystander effect after ionizing radiation, manifested as increased yield of micronuclei was eliminated by treating the cells with iNOS inhibitor, aminoguanidine, or anti-TGF-beta1 antibodies. Additionaly, intracellular level of NO was increased in bystander cells, indicating NO as a mediator of the bystander effect (Shao et al., 2008a). However, in our UVR experiments (Widel et al., 2014) NO played a minor role, since its level was almost unchanged in irradiated and bystander cells.
We also observed an increase in IL-6 in the medium collected from the culture of cells exposed to all three UV bands and incubated alone, or co-incubated with unexposed cells (Widel et al., 2014). The level of IL-6 in the medium obtained in an experiment with co-incubation was higher than in medium collected from irradiated cells incubated alone, thus IL-6 had to be also generated by the non-irradiated cells. Senescent cells probably secreted this cytokine, since the increase of IL-6 observed in the co-incubation system after UVC and UVB exposure was associated with senescence induction in bystander cells.
These findings support the hypothesis that damage in bystander cells results from their exposure to reactive oxygen compounds and proinflamatory cytokines released by UVR-stressed cells.
Protective Bystander Effect
Though bystander effect is generally detrimental to the cells, some studies show its protective action. The soluble molecules released to the medium by A375 human melanoma cells exposed to UVC exerted a protective effect for the cells subsequently exposed to UVC or hydrogen peroxide (Ghosh et al., 2012, 2013). This effect can be termed an adaptive response, similar to the ionizing radiation-induced adaptive response (Azzam et al., 1994). Increasing proliferation of A375 cells was associated with stimulated antioxidant defense via an increase in CAT and cytoplasmic SOD activity, whereas glutathione peroxidase and GSH levels remained unaffected.
A process of melanogenesis stimulation found in B16 melanoma cells neighboring the cells exposed to both, UVA and UVB radiation (Nishiura et al., 2012) may also be considered as a protective bystander effect. Because the melanin produced by melanocytes is a natural skin protective agent against UV radiation, by scavenging reactive oxygen species (Chedekel et al., 1978), a protective bystander effect seems obvious after melanocyte exposure to UV radiation. However, melanogenesis in the Nishiura study was accompanied by ROS generation in bystander cells, followed by a decline of mitochondrial membrane potential, the markers of damaged cells. The impairment of mitochondrial membrane potential was associated with an influx of calcium ions in the UVA system, but not in the UVB system. These results indicate that melanogenesis in bystander cells proceedes via different pathways in the UVA and UVB systems. The differences are in some accordance with the data of Kvam and Dahle (2004), which showed that melanin sensitized melanocytes to oxidative DNA damage by UVA radiation, but protected them from direct DNA damage by UVB radiation.
Recently, Redmond et al. (2014) showed that UVA-exposed dermal cells (keratinocytes, melanocytes, and fibroblasts) induced intercellular oxidative signaling to non-irradiated bystander cells, but melanocytes were the most susceptible to bystander oxidative signaling. Thus, in melanocytes exposed to UVA, two events, melanogenesis and the increase of oxygen reactive species may operate oppositely with unpredictable final consequences.
Conclusions
The studies published so far show that the bystander effects are induced in the cells of both normal and neoplastic types after exposure to UVA, UVB and UVC radiation. These effects are disclosed in neighboring cells as well as daughter cells (genetic instability), and are generally detrimental. Cellular effects in bystander cells such as: lowering of survival, induction of apoptosis, micronuclei, chromosomal aberrations and mutagenicity elicit with varying degrees and different kinetics after exposure to different UV bands. Premature senescence in response to oxidative stress generated by UV radiation is also observed in exposed and bystander cells. Sometimes the UV-induced bystander effect has a protective character. The majority of published data show that all three UV bands are efficient inducers of damaging effects not only in UV-exposed but in bystander cells through the generation of ROS and proinflamatory cytokines. Although further studies are required to better understand the nature of the mediators of UV-induced bystander effects, but nonetheless available data suggest that all three UVR bands carry a potential health risk not only due to direct mechanisms, but also through the bystander phenomenon.
References
Alexandre J, Hu Y, Lu W, Pelicano H, Huang P. 2007. Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res 67:3512-3517.
Azzam EI, Raaphorst GP, Mitchel RE. 1994. Radiation-induced adaptive response for protection against micronucleus formation and neoplastic transformation in C3H 10T1/2 mouse embryo cells. Radiat Res 138(1 Suppl):S28-31.
Banerjee G, Gupta N, Kapoor A, Raman G: UV induced bystander signaling leading to apoptosis. Cancer Lett 223:275-284.
Chakraborty A, Held KD, Prise KM, Liberd HL, and Redmond RW.2009. Bystander effects induced by diffusing mediators after photodynamic stress. Radiat Res 172:74-81.
Chedekel MR, Smith SK, Post PW, Pokora A, and Vessell DL. 1978. Photodestruction of pheomelanin: role of oxygen. Proc Natl Acad Sci USA 75:5395-5399.
Dahle J, Bagdonas S, Kaalhus O, Olsen G, Steen HB, Moan J. 2000. The bystander effect in photodynamic inactivation of cells. Biochim Biophys Acta 1475;273-280.
Dahle J, Kvam E. 2003. Induction of delayed mutations and chromosomal instability in fibroblasts after UVA-, UVB-, and X-radiation. Cancer Res 63:1464-1469.
Dahle J, Kaalhus O, Stokke T, Kvam E. 2005a. Bystander effects may modulate ultraviolet A and B radiation induced delayed mutagenesis. Radiat Res 163:289-295.
Dahle J, Kvam E, Stokke T. 2005b. Bystander effects in UV-induced genomic instability: antioxidants inhibit delayed mutagenesis induced by ultraviolet A and B radiation. J Carcinog 4:11-19.
de Gruijl FR, van der Leun JC. 1994. Estimate of the wavelength dependency of ultraviolet carcinogenesis in humans and its relevance to the risk assessment of stratospheric ozone depletion. Health Phys 67:319-325.
Dickey J S, Baird B J, Redon CE, Sokolov MV, Sedelnikova OA and Bonner WM. 2009. Intercellular communication of cellular stress monitored by γ-H2AX induction. Carcinogenesis 30:1686-1695.
Esterbauer H, Schaur RJ., Zollner H. 1991. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol. Med 11: 81-128.
Harada T, Kashino G, Suzuki K, Matsuda N, Kodama S, Watanabe M. 2008. Different involvement of radical species in irradiated and bystander cells. Int J Radiat Biol 84:809-814.
Hockberger PE. 2002. A history of ultraviolet photobiology for humans, animals and microorganisms. Photochem Photobiol 76:561-579.
Kadhim MA, Lorimore SA, Townsend KM, Goodhead DT, Buckle V, Wright E G. 1995. Radiation induced genomic instability: Delayed cytogenetic aberrations and apoptosis in primary human bone marrow cells. Int J Radiat Biol 67:287-293.
Kulms D, Zeise E, Pöppelmann B, and Schwarz T. 2002. DNA damage, death receptor activation and reactive oxygen species contribute to ultraviolet radiation-induced apoptosis in an essential and independent way. Oncogene 21:5844-5851.
Kvam E, Dahle J. 2004. Melanin synthesis may sensitize melanocytes to oxidative DNA damage by ultraviolet A radiation and protect melanocytes from direct DNA damage by ultraviolet B radiation. Pigment Cell Res 17:549-550.
Marnett LJ. 2000. Oxyradicals and DNA damage. Carcinogenesis 21:361-370.
Masaki H, Atsumi I, Sakurai H. 1995. Detection of hydrogen peroxide and hydroxyl radicals in murine skin fibroblasts under UVB irradiation. Biochem Biophys Res Commun 206:474-479.
McMillan TJ, Leatherman E, Ridley A, Shorrocks J, Tobi SE and Whiteside JR. 2008. Cellular effects of long wavelength UV light (UVA) in mammalian cells. J Pharm Pharmacol 60:969-976.
Morliere P, Moysan A, Tirache I. 1995. Action spectrum for UV-induced lipid peroxidation in cultured human skin fibroblasts. Free Radic Biol Med 19:365-371.
Mothersill C, Seymour CB. 2012. Are epigenetic mechanisms involved in radiation-induced bystander effect? Front Genet 3: 74.
Nishiura H, Kumagai J, Kashino G, Okada T, Watanabe M. 2012. The bystander effect is a novel mechanism of UVA induced melanogenesis. Photochem Photobiol 88:389-397.
O'Reilly JP, Mothersill C. 1997. Comparative effects of UV A and UV B on clonogenic survival and delayed cell death in skin cell lines from humans and fish. Int J Radiat Biol 72:111-119.
Phillipson RP, Tobi SE, Morris JA, McMillan TJ. 2002. UV-A induces persistent genomic instability in human keratinocytes through an oxidative stress mechanism. Free Radic Biol Med 32:474-480.
Prise KM, Folkard M, Michael BD. 2003. Bystander responses induced by low LET radiation. Oncogene 22:7043-7049.
Rastogi RP, Richa, Kumar A, Tyagi MB., Sinha RP. 2010. Molecular Mechanisms of Ultraviolet Radiation-Induced DNA Damage and Repair. J Nucleic Acids Article ID 592980.
Redmond RW, Rajadurai A, Udayakumar D, Sviderskaya EV, Tsao H. 2014. Melanocytes are selectively vulnerable to UVA-mediated bystander oxidative signaling. J Invest Dermatol 134:1083-1090.
Rünger TM. 2007. How different wavelengths of the ultraviolet spectrum contribute to skin carcinogenesis: The role of cellular damage responses. J Invest Dermatol 127:2103-2105.
Rzeszowska-Wolny J, Przybyszewski WM, Widel M. 2009. Ionizing radiation induced bystander effects, potential targets for modulation of radiotherapy. Eur J Pharmacol 625:156-167.
Setlow RB, Grist E, Thompson K, Woodhead AD. 1993. Wavelengths effective in induction of malignant melanoma. Proc Natl Acad Sci USA 90: 6666-6670.
Shao C, Folkard M, Prise KM. 2008. Role of TGF-beta1 and nitric oxide in the bystander response of irradiated glioma cells. Oncogene 27:434-440.
Slieman TA; Nicholson WL. 2000. Artificial and solar UV radiation induces strand breaks and cyclobutane pyrimidine dimers in Bacillus subtilis spore DNA. Appl Environ Microbiol 66:199-205.
Suzuki M, Boothman DA. 2008. Stress-induced premature senescence (SIPS) - influence of SIPS on radiotherapy. J Radiat Res 49:105-112.
Wang TC, Smith KC. 1986. Postreplication repair in ultraviolet-irradiated human fibroblasts: formation and repair of DNA double-strand breaks. Carcinogenesis 7:389-392.
Whiteside JR, Allinson SL, McMillan TJ. 2011. Timeframes of UVA-induced bystander effects in human keratinocytes. Photochem Photobiol 87:435-440.
Whiteside JR, McMillan TJ. 2009. A bystander effect is induced in human cells treated with UVA radiation but not UVB radiation. Radiat Res 171:204-211.
Widel M. 2012. Bystander effect induced by UV radiation; why should we be interested? Postepy Hig Med Dosw (Online) 66:828-837.
Widel M, Krzywon A, Gajda K, Skonieczna M, Rzeszowska-Wolny J. 2014. Induction of bystander effects by UVA, UVB, and UVC radiation in human fibroblasts and the implication of reactive oxygen species. Free Radic Biol Med. 68:278-287.
Widel M, Przybyszewski WM, Cieslar-Pobuda A, Saenko YV, Rzeszowska-Wolny J. 2012. Bystander normal human fibroblasts reduce damage response in radiation targeted cancer cells through intercellular ROS level modulation. Mutat Res 731:117-124.
Yoshizumi M, Nakamura T, Kato M, Ishioka T, Kozawa K, Wakamatsu K, Kimura H. 2008. Release of cytokines/chemokines and cell death in UVB-irradiated human keratinocytes, HaCaT. Cell Biol Int 32:1405-1411.
Zhong W, He Q, Chan LL, Zhou F, El Naghy M, Thompson EB, Ansari NH. 2001. Involvement of caspases in 4-hydroxy-alkenal-induced apoptosis in human leukemic cells. Free Rad Biol Med 30:699-706.
09/08/14