HUMAN PHOTOSENSITIVE DISEASES OF DNA REPAIR
James E. Cleaver
Auerback Melanoma Laboratory
Box 0808, UCSF Cancer Center
University of California
San Francisco, CA 94143-0808
(415) 476-4563
james.cleaver@ucsf.edu
DNA Damage From Ultraviolet Light
Ultraviolet radiation (UVR) covers three wavelength ranges: UVA (320-400 nm), UVB (290-320 nm), and UVC (240-290 nm). UVA is photocarcinogenic and is involved in photoaging, but is weakly absorbed in DNA and protein, and is absorbed by other chromophores that lead to reactive oxygen species (ROS), which secondarily cause damage to DNA (Tyrrell and Keyse, 1990). UVB overlaps the upper end of the DNA and protein absorption spectra, is present in sunlight, and contains the wavelengths mainly responsible for skin cancer through direct photochemical damage to DNA. UVC is not present in sunlight, but is readily produced by low-pressure mercury sterilizing lamps (254 nm), which coincides with the peak of DNA absorption (260 nm). These lamps and are used extensively in experimental studies. The absorption of UV by stratospheric ozone results in negligible radiation shorter than 300 nm reaching the earth's surface.
The proliferating cells in the basal layer of the skin epithelium are protected by melanin pigment and keratin layers; intracellular defenses depend upon repair of the DNA damage, antioxidant enzymes (superoxide dismutase, catalase, glutathione reductase, etc.), endogenous free radical quenchers, and inducible detoxifying enzymes and biochemical systems (Tyrrell and Keyse, 1990). Although the important wavelength range responsible for skin cancer is UVB, UVA cannot be excluded because of evidence that it may indirectly form thymine dimers in skin (Courdavault et al., 2004; Mouret et al., 2006), and is carcinogenic in mice (Sterenborg and van der Leun, 1990).
Sunlight-Induced Photoproducts in DNA
The absorption spectrum of DNA correlates well with UV-induced lethality, mutation, and photoproduct formation (Niggli and Cerutti, 1983; Jones et al., 1987; Tyrrell and Pidoux, 1987; Pfeifer et al., 1991, 1992). Dimerizations between adjacent pyrimidines are the most prevalent photoproducts found in DNA. Most important are the cyclobutane pyrimidine dimer (CPD) and, at about 25% the frequency, the [6-4] pyrimidine dimer [(6-4)PD], which are preferentially induced at thymine-cytosine dipyrimidines (Mitchell and Cleaver, 1990). The relative proportion of DNA photoproducts varies across the UV spectrum. The distribution of these photoproducts in human chromatin and their repair depends on base sequence, secondary DNA structure, and DNA-protein interactions (Mitchell et al., 1991, 1992; Pfeifer et al., 1992; Lange et al., 2008). Cytosine absorbs longer wavelengths of UV more efficiently than thymine, and therefore CPDs containing cytosine are formed more readily after UVB irradiation (Ellison and Childs, 1981), and may play a major role in UVB (solar) mutagenesis. The [6-4]PD can undergo a UVB-dependent conversion to its valence photoisomer, the Dewar pyrimidinone (Taylor and Cohrs, 1987). Other less common lesions include purine-purine and purine-pyrimidine photoadducts, photohydrations, and photooxidations (Cadet and Vigney, 1990).
Mutations induced by UV radiation usually bear the footprints of pyrimidine dimers that caused them. They most often occur where cytosine is a component of the photoproduct, since the insertion of adenine opposite thymine is a correct and non-mutagenic event. Hence, many CPDs that form between two thymine bases are not mutagenic, but T=C dimers result in C to T mutations at the cytosine site. Since the [6-4]PD is considerably more distortive than the CPD (i.e., it causes a 47o as opposed to a 7o helical bend) it is more likely to block DNA synthesis and be lethal than mutagenic. Conversely the distortion results in the [6-4]PD being more rapidly recognized and excised during repair. Because damage bypass and base insertion depend on a variety of conditions, both CPDs and (6-4)PDs contribute to mutagenesis and tumorigenesis in a complex manner.
UVA primarily produces damage indirectly through ROS, which can be reduced by free radical scavengers (Tyrrell and Pidoux, 1986). ROS can react with DNA to form base damage, strand breaks and DNA-protein crosslinks that may be an important pathogenic component of sunlight (Jones et al. 1987; Tyrrell and Pidoux, 1987) (see module on DNA-Protein Crosslinks). Recent evidence suggests that UVA may also induce significant levels of CPDs in human cells by photosensitized triplet energy transfer, and that these lesions should be taken into account to fully understand the biological effects of UVA (Courdavault et al., 2004).
Photosensitive and DNA Repair Deficient Diseases
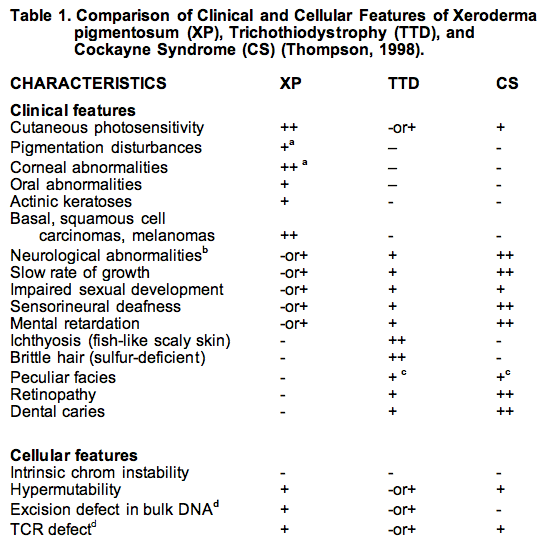
Xeroderma pigmentosum (XP) was the first nucleotide excision repair (NER) disease to be identified (Cleaver, 1968, 1969) (see module on Nucleotide Excision Repair). This discovery linked a human mutation to solar–sensitivity and cancer, and provided an impetus to the concept of somatic mutations and genomic instability as a causative factor in human cancer. The complete family of NER-related diseases now include (Figure 1, Table 1): XP itself, XP with neurological complications (originally called the de Sanctis-Cacchione syndrome), the XP variant, Cockayne syndrome (CS), the cerebro-oculo-facio-skeletal syndrome (COFS), a mild UV-sensitive syndrome (UVs), trichothiodystrophy (TTD), and patients with combined symptoms of XP/CS and XP/TTD (Kraemer et al., 1987; Bootsma et al., 1998; Thompson, 1998; Cleaver and Crowley, 2002). These diseases show overlapping symptoms associated with cancer, developmental delay, immunological defects, neurodegeneration, retinal degeneration, premature aging, and impairment of the growth hormone-insulin-like growth factor 1 (IGF-1) axis (van der Pluijm et al., 2006).
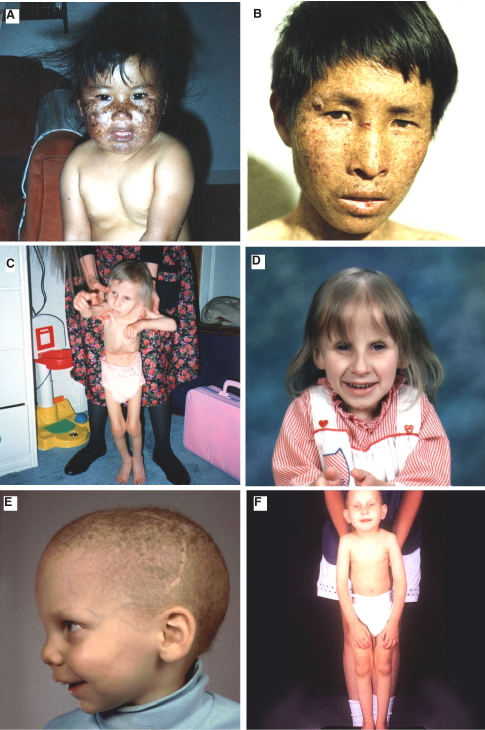
Figure. 1. (A,B) Two XP patients of undefined complementation group; the patient in panel A subsequently showed decline of the central nervous system. (C,D) A CS patient at ages 4 and almost 7 yr, respectively. (E,F) two TTD patients. [Reproduced from Thompson, 1998.]
Reproduced from Thompson (1998) with the permission of Springer Science and Business Media.
a. Traits that are hallmarks of the disease are indicated by (++), and traits that are sometimes associated with the disorder are indicated by (+); unassociated traits are shown by (-).
b. These features are present in about 20% of XP patients. The molecular basis of the neurological dysfunctions in XP differs from that of CS and TTD. Demyelination of neurons is seen in TTD and CS. XP involves primary neuronal degeneration thought to be related to the severity of repair deficiency and metabolic damage to DNA.
c. The facial abnormalities of TTD and CS overlap in term of protruding ears. TTD often has receding chin and small thin or beaked nose, whereas CS tends to have large nose and projecting jaw.
d. XPC is exceptional in being deficient in repairing bulk DNA, but proficient in transcription coupled repair (TCR).
Xeroderma Pigmentosum
Xeroderma pigmentosum (XP) (Cleaver, 1968) is a rare human, autosomally inherited, skin and neurodegenerative disease in which exposure to normal sunlight can result in a very high incidence of skin and mucous membrane cancer. The disease occurs at a frequency of 2.3 per million in Western Europe, including both indigenous and immigrant populations; when these are segregated the rate is 0.9 per million for indigenous European populations (Kleijer et al., 2008). Heterozygotes are unaffected, in contrast to the severity of symptoms in the homozygotes (Kraemer et al., 1987, 1994). UVB is the shorter wavelength radiation in sunlight responsible for most sun-induced cancers in the general population, as well as in XP patients. The symptoms of XP begin in early life with the first exposures to sunlight, the median age of onset being 1–2 years of age, with skin rapidly exhibiting the signs associated with years of sun exposure. Some patients present with severe sunburn and blistering; others present with early extensive freckling without severe sunburn. Patients from XP groups C and V rarely burn, but their skin cancers emerge earlier than other groups that burn and blister severely (Bradford et al., 2011). Pigmentation is patchy, and skin shows atrophy and telangiectasia with the development of basal and squamous cell carcinomas and melanomas. In a survey of clinical symptoms coupled to genotypes of XP patients in England, patients who displayed severe sunburns developed their skin cancers later with lower frequency than those with extensive freckling (Fessihi et al., 2016). Some patients in this group showed widely different clinical presentations despite having the identical mutations in an XP gene.
The relative incidence of the various forms of skin cancers in XP patients, including nonmelanoma skin cancers (NMSC) and melanomas, is similar to that in the general population (Kraemer et al., 1994). In a summary of 106 patients, cancer incidence for those individuals under 20 years of age was 10,000 times that seen in the general population for NMSC, and 2,000 times for melanoma (Bradford et al., 2011). The median age at diagnosis of first non-melanoma skin cancer was 9 years, younger than the median age of 22 years for melanoma. This is a reversal from the general population in which NMSC generally occurs later in life than melanoma. This reversal of age of incidence suggests a more prominent role for solar exposure and DNA repair defects for NMSC, in comparison to melanoma in XP patients. Progressive neurologic degeneration occurs in a significant number of patients, which can be correlated with mutations in specific XP genes (XPA, B, D, G). The causes of death include skin cancer (34%), neurological degeneration (31%), and internal cancer, with the median age at death in XP patients with neurodegeneration being 29 years; those without neurodegeneration 37 years (Bradford et al., 2011). A specific effect on lifespan of XP cells in culture is not, however, apparent (Cleaver, 1984).
The specificity of cytosine-containing CPDs in mutagenesis is exemplified in each of the three main types of skin cancer in XP patients, where driver mutations occur at higher rates than in non-XP patients. In melanoma, activating mutations occur in the promoter of the telomerase gene hTERT (Horn et al., 2013; Huang et al., 2013), and inactivating mutations in the PTEN gene (Wang et al. 2009) at CPD sites. The common BRAF gene mutation (V600E) in melanoma, however does not occur at CPD sites (Maldonado et al., 2003), and is likely caused by mechanisms other than sun exposure (Mitra et al., 2012). In squamous cell carcinoma two common mutations involve the p53 and NOTCH1 genes, and both show UV-type mutations (Wang et al., 2011). Similar UV-type mutations occur in the causative genes, Ptch1 and Smo, in basal cell carcinomas; Smo mutations occur uniquely in XP (Couve-Privat et al., 2002).
There has been some question of whether XP patients show a higher than normal incidence of internal cancers (Kraemer et al., 1984), but answers are bedeviled by small sample sizes and the possibility that the low normal levels of vitamin D associated with sun protection of XP patients (Sollitto et al., 1997) could influence the internal cancer rates (Garland and Garland, 1980; Dixon et al., 2005; Spina et al., 2006). The summary of 106 patients from NIH reported 3 central nervous system cancers and a peripheral nerve cancer, plus 2 other internal cancers (Bradford et al., 2011).
Cockayne Syndrome (CS)
CS is an autosomal recessive disease characterized by cachectic dwarfism, retinopathy, microcephaly, deafness, neural defects, and retardation of growth and development after birth (Figure 1) (Nance and Berry, 1992). The cerebellum shows a distinctive loss of Purkinje cells associated with regulating balance and walking. They have a typical facial appearance with sunken eyes and a beaked nose and projecting jaw. CS patients are sun sensitive, but have never been reported to develop skin cancers, setting this disease apart from XP (Nance & Berry, 1992; Zhang et al., 2016). CS patients carry mutations in one of two genes, CSA or CSB, group B being the more common (Laugel et al., 2010). In addition there are patients who show combined XP and XP/CS symptoms, some of whom have mutations in XPB, XPD or XPG (Weeda et al., 1990). The CS gene products are involved in coupling excision repair to transcription (Hoeijmakers, 2001). They may be involved in the ubiquitination and degradation of stalled RNA pol II at damaged sites, and activation of ATM-dependent signaling (Bregman et al., 1996; Groisman et al., 2003; Tresini et al., 2015). The CS gene products have an additional role in mitochondrial functions that may be important for the developmental and neurodegenerative symptoms (Aamann et al., 2010; Scheibye-Knudsen et al., 2012; Cleaver et al., 2014).
Cockayne syndrome may be considered to encompass a wide range of disease from the mild UVs syndrome that only shows photosensitivity, through the major groups known clinically as CS I, II and III, through to the most severe neonatal lethal disorder Cerebro-Oculo-Facio Skeletal (COFS) syndrome. The majority of patients within the classic type I do not survive beyond the second decade of life (Laugel et al., 2008a). Type II is more severe with a shorter life-span, and type III is a milder form with a longer life span. No correlation has been established between the genes, their mutations and the clinical presentations (Laugel et al., 2008a). The absence of skin cancer, despite the high degree of photosensitivity, is due to the functions of the transcription coupled repair (TCR) system affected in CS (see module Nucleotide Excision Repair in Human Cells). The CSA or CSB mutations interrupt TCR resulting in increased cytotoxicity but no increase in UV-induced mutations (Reid-Bayless et al., 2016). This is in contrast to XP where the repair deficiency increases both UV-induced mutagenesis and skin cancer (Maher et al.,1979).
Trichothiodystrophy (TTD)
TTD is a rare autosomal recessive disorder characterized by sulfur-deficient brittle hair and ichthyosis (Figure 1) (Itin et al., 2001). Hair shafts split longitudinally into small fibers, and this brittleness is associated with levels of cysteine/cystine in hair proteins that are 15 to 50% of those in normal individuals. The hair has characteristic "tiger-tail" banding visible under polarized light. The patients often have an unusual facial appearance, with protruding ears and a receding chin. Mental abilities range from low normal to severe retardation (Lehmann et al., 1988).
A recent systematic literature review (Faghri et al., 2008) identified 112 patients ranging from 12 weeks of age to 47 years (median 6 years). In addition to hair abnormalities, common features reported were developmental delay/intellectual impairment (86%), short stature (73%), ichthyosis (65%), abnormal physical characteristics at birth (55%), ocular abnormalities (51%), infections (46%), photosensitivity (42%), maternal pregnancy complications (28%) and defective DNA repair (37%). There was high mortality, with 19 deaths under age 10 years (13 infection-related), which is 20-fold higher compared to the US population. The spectrum of clinical features varied from mild disease with only hair involvement to severe disease with profound developmental defects, recurrent infections and a high mortality at a young age (Faghri et al., 2008). Abnormal characteristics at birth and pregnancy complications, unrecognized but common features of TTD, suggest a role for DNA repair genes in normal fetal development.
Several categories of the disease can be recognized on the basis of cellular responses to UV damage and the affected genes. Severe photosensitive and NER defective cases of TTD cases involve components of the transcription factor TFIIH (Broughton et al., 1990; Itin et al., 2001): XPB, XPD or a small stabilizing factor p8 (Giglia-Mari et al., 2004; Ranish et al., 2004). There are also nonphotosensitive TTD cases that involve at least one more gene, TTDN1 (Botta et al., 2007). Although TTD patients do not exhibit increased incidence of skin cancer, corresponding mice with a human TTD mutation do demonstrate increased UV-induced skin cancer, indicating important differences between the human and mouse models (de Boer et al., 1998a, b).
XP-CS and XP-TTD Syndrome
There are several examples of patients who exhibit combined symptoms of XP and other developmental and neurological disorders of CS or TTD. These have generally been found to correspond to mutations in the XPB, XPD or XPG genes.
Cerebro-Oculo-Facio Skeletal (COFS) syndrome
Cerebro-Oculo-Facio-Skeletal (COFS) syndrome is an autosomal, recessively inherited and rapidly progressive, neurologic disorder. The disease leads to brain microcephaly and atrophy with calcifications, cataracts, microcornea, optic atrophy, progressive joint contractures, and growth failure. COFS appears to be a particularly severe developmental and neurological expression of mutations in CSB, XPG, XPD and ERCC1 (Graham et al., 2001; Niedernhofer et al., 2006; Jaspers et al., 2007; Laugel et al., 2008b).
The XP, CS and TTD Genes
Eight genes have been identified among XP patients (Thompson, 1998) (Table 2); seven are involved in nucleotide excision repair (XPA-XPG) and one, the XP variant, is involved in replication of damaged DNA on the leading strand (Svoboda et al., 1998). Mutations in any of the genes XPA to XPG result in reductions in the ability of cells to excise cyclobutane pyrimidine dimers, [6–4] photoproducts, and other bulky carcinogen adducts from DNA. The functions of most of the gene products in the NER process have been identified (for greater detail see module on Nucleotide Excision Repair in Human Cells), and mutations located in each of the genes can in some cases be correlated with cellular functions and with severity of disease (Hoeijmakers, 2001; Cleaver and Crowley, 2002; Cleaver, 2004, 2005). Several of the gene products occur in heterodimeric complexes with proteins that are essential for their stability and function: the XPC protein is complexed with HHR23B; the 48 kDa XPE protein (DDB2) is complexed with a larger 127 kDa subunit (DDB1); and the XPF protein is complexed with the ERCC1 protein (ERCC = excision repair cross complementing, to indicate a human gene correcting a rodent cell mutation). Curiously, in each of these examples, mutations associated with XP have mainly been found in one of the members of the heterodimer. In addition, the XPB and XPD helicases exist as components of TFIIH, which is a basal transcription initiation factor containing at least ten proteins (Hoeijmakers et al., 1996).
XPA
XPA is located on chromosome 9q34.1, and encodes a 273 amino acid Zn2+ finger protein (XPA) that participates in photoproduct recognition and DNA binding, sometimes known as a verification step (Miura et al., 1991; Jones and Wood, 1993; Asahina et al., 1994). XPA acts subsequent to the earliest damage recognition steps that involve DDB2, the protein product of the XPE gene, and XPC (Kapetanaki et al., 2006; Scrima et al., 2009; Robu et al., 2013). XPA binding may be followed by the formation of a quasi-stable complex consisting of XPA, XPC, human single-strand binding protein (RPA/HSSB), and TFIIH, which then acts as a nucleation site for binding of the incision/excision enzymes (Mu et al. 1997). Of the two major photoproducts, [6–4] photoproducts and cyclobutane pyrimidine dimers, XPA alone shows only weak binding to CPDs (Miura et al., 1991; Robins et al. ,1991; Jones and Wood, 1993; He et al., 1995; Li et al., 1995b, c). The XPA-RPA complex appears to bind to damaged sites in DNA once they have been recognized and bound by either XPC/HHR23B in nontranscribed regions of DNA (Sugasawa et al., 1998), or by stalled RNA polymerase II transcription machinery in transcribed regions (Hanawalt, 1994). XPA is also required for the repair of oxidative damage in mitochondrial DNA (Driggers et al., 1996), indicating a potential overlap between nucleotide and base excision repair, also found with XPG (Cooper et al., 1997).
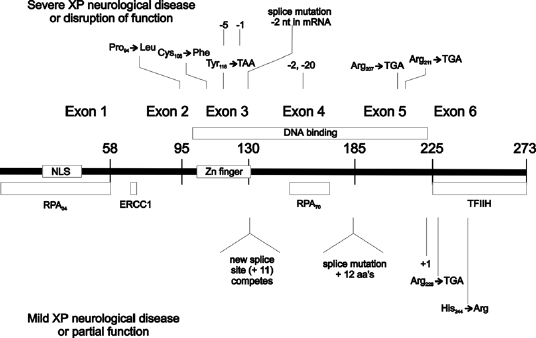
Figure 2. Location of the mutations in the XPA gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Reproduced from Figure 6 in States et al., Human Mutation 12:103-113, 1998, with permission.]
The gene is encoded in 6 exons distributed over 22–25 kb of genomic DNA (Satokata et al., 1993; Topping et al., 1995), and mutations have been found throughout the gene with the exception of exon I. The first exon is essential for nuclear localization, but not for DNA repair when the protein is expressed at high levels (Miyamoto et al., 1992). Exon II encodes a domain for binding to ERCC1, a component of the heterodimeric 5' endonuclease composed of ERCC1 and XPF (ERCC4) (Li et al., 1995a, b), and deletion of the ERCC1 binding region in vitro generates a dominant negative phenotype (Li et al., 1995b). Exon III encodes the Zn2+ finger that binds RPA (Li et al., 1995a). Exons IV and V comprise the DNA binding domain (Kuraoka et al., 1996; Ikegami et al., 1998). Exon VI interacts with the TFIIH complex (Park et al., 1995).
Two classes of XP-A patients are known, those with severe CNS disorders involving sensoneural deafness, reduced nerve conduction, difficulty walking, and occasionally microcephaly, and those with only skin sensitivity and cancer. The more severe cases tend to have both alleles with mutations that occur within the DNA binding region of the protein, often resulting in truncation, and detailed analysis and diagrammatic representation has been published previously (States et al., 1998). Severe cases in both Japan and Africa have been reported with mutations at the same site, the last nucleotide of intron 3. Milder cases generally have at least one allele with a mutation outside of the DNA binding region, such as the exon 6 (R228ter) mutation common in Tunisian patients (Nishigori et al., 1993, 1994), and other cases (Cleaver et al., 1995). A few other mild cases have mutations close to a splice site such that alternative splicing may allow the persistence of a low level of normal protein.
XPB (ERCC3)
XPB is located on chromosome 2q21, and encodes a 782 amino acid 3'–5' helicase, the p89 subunit of TFIIH (Coin et al., 1999). The gene is also known as ERCC3. The helicase may be involved in unwinding the DNA 5'-ward of a damaged base. The ATPase activity is required to "lock" the pre-incision complex in place prior to cleavage of the DNA strands (Wakasugi and Sancar, 1999; Reardon and Sancar, 2005; Coin et al., 2007; Oh et al., 2007). The N terminus of the protein interacts with the XPD and XPG proteins, whereas the C terminus is required for the 5' cleavage during excision repair (Evans et al., 1997). The protein also interacts with the BCR-ABL oncoprotein, although its potential role in hematopoeitic malignancies is unknown (Takeda et al., 1999). Only a small number of patients are known in this complementation group, and their clinical symptoms are extremely varied with mutations at both extremes of the gene.

Figure 3. Location of the mutations in the XPB gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Reproduced from Itin et al., J. Amer. Acad. Dermatology 44:891-920, 2001, with permission from Mosby Inc.]
XPC
XPC is located on chromosome 3p25.1, and encodes a 940 amino acid single-stranded DNA binding protein that is essential for the repair of the nontranscribed regions of the genome. XPC patients are among the more common form of XP, and exhibit predominantly skin cancer without neurological disorders, although rare cases with neurologic symptoms have been reported. The protein acts in an early step of damage recognition in nontranscribed regions (Sugasawa et al., 1998; Wang et al., 2004), but is released from the pre-incision complex before strand incision occurs (Wakasugi and Sancar, 1998), a process that involves ubiquitylation by the DDB1/2 (XPE) ubiquitin ligase (Sugasawa et al., 2005). XPC exists in vivo in a tight complex with another protein HHR23B, which is encoded by a closely linked (within 650 kb) gene to XPC (Masutani et al., 1994). Mutations in HHR23B are not known to be associated with any human disorder, although the mutations in the corresponding gene in yeast makes cells UV sensitive.
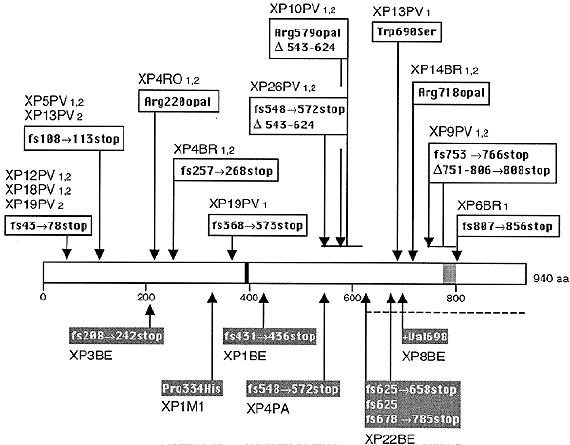
Figure 4. Location of the mutations in the XPC gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Reproduced from Figure 6 in Chavanne et al., Cancer Research 60: 1974-1982, 2001, with permission.]
XPD (ERCC2)
The XPD gene is located on chromosome 19q13.2, and encodes a 760 amino acid 5'–3' helicase, a component of transcription factor TFIIH (Bootsma et al., 1998; Coin et al. 1998, 1999). The gene, encoded in 23 exons, is also known as ERCC2. The helicase may be involved in 3'-ward unwinding of the DNA in the vicinity of a damaged base, and in the opposite direction to the XPB helicase. The phenotypes of mutations in this gene are among the most complex of all XP groups, being associated with three different clinical disorders: XP group D, TTD, or a rare combination of XP and Cockayne syndrome. Most TTD patients exhibiting UV sensitivity fall into the XP-D complementation group (Stefanini et al., 1993; Botta et al., 1998).
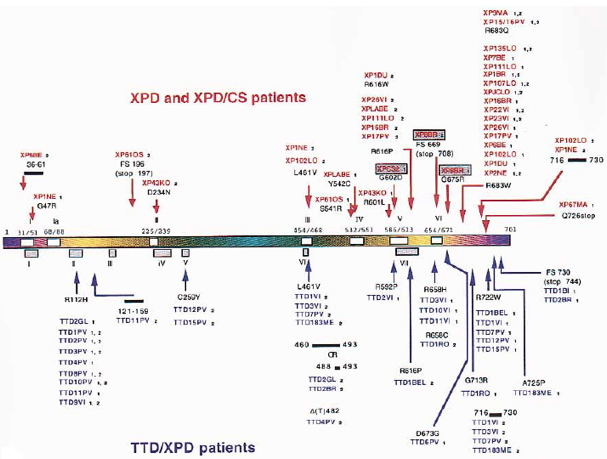
Figure 5. Location of the mutations in the XPD gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. The mutations associated with the three diseases XP, CS and TTD are identified by different colors. [Reproduced from Itin et al., J. Amer. Acad. Dermatology 44:891-920, 2001, with permission from Mosby Inc.]
Since the clinical characteristics of XP and TTD are so different, mutation analysis has sought to explain how two dissimilar disorders are associated with the same gene. The result of such studies is that all mutations thus far reported appear to be specific for the disease, and only missense mutations are important for disease, because loss of function mutations are lethal, although the severity can be influenced by gene dosage (Botta et al., 1998). As a member of the TFIIH complex, XPD is an essential protein for transcription and cell viability (de Boer et al., 1998b). Thus, no patient can have two null alleles. The interpretation of XPD mutations is confounded by the fact that in several cases XPD and TTD patients have an allele in common, suggesting that the second alleles determine the clinical presentation. By examining the phenotype in Schizosaccharomyces pombe of mutations homologous to those present in these shared alleles, the S. pombe mutations were shown to behave as null mutations (Taylor et al., 1997).
Therefore, by extrapolation, these common alleles can conveniently be regarded as nonfunctional and playing no direct role in the specific disease. Examination of the distribution of mutations within the XPD protein reveals that essentially all XPD mutations fall within one of the conserved helicase domains (Koonin, 1993). This pattern indicates that these mutations can be expected to reduce the protein’s helicase activity (Coin et al., 1998). In contrast, the TTD mutations usually fall outside of the helicase domains, and show significant clustering at the C-terminus of the protein. TTD-specific mutations may subtly interfere with the ability of XPD to interact with its partner proteins within the TFIIH complex, and thereby destabilize the complex (Coin et al., 1998) and cause subtle deficiencies in transcription (Hoeijmakers et al., 1996; de Boer et al., 1998a). Alternatively, the TTD mutations affect recruitment of the TFIIH complex to cyclobutane dimers, but not to [6-4] photoproducts (Chiganças et al., 2008).
XPE (DDB1 /DDB2)
The XP-E complementation group includes patients who are mildly to moderately affected, and whose cells carry out near normal levels of NER (Itoh et al., 2000). Since excision repair is close to normal in XPE cells, a definitive diagnosis of XPE requires sequencing of the XPE gene (DDB2) to identify mutations (Nichols et al., 1996; Cleaver et al., 1999; Itoh et al. 2000, 2001).
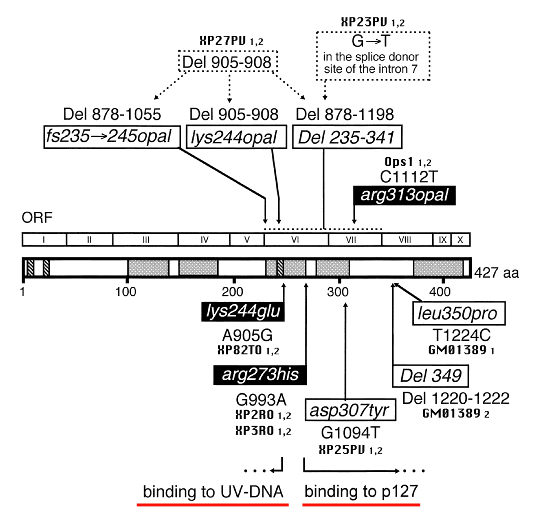
Figure 6. Location of the mutations in the XPE (DDB2) gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Figure provided by Dr. Toshiko Itoh, UC Berkeley.]
The XPE group involves mutations in one component of a dimeric protein having subunits of 127 kDa (DDB1) and 48 kDa (DDB2) (Chu and Chang, 1988; Keeney et al., 1993; Nichols et al., 1996, 2001). The DDB1 and DDB2 genes are located on chromosome 11q12-13 and 11p11-12, respectively (Dualan et al., 1995). Mutations diagnostic for XPE patients are located in the DDB2 subunit and mildly affected patients without such mutations are assigned to other groups after further diagnosis (Nichols et al., 1996; Cleaver et al., 1999; Itoh et al. 2000, 2001). The DDB1/2 heterodimer is involved with the initial recognition of UV-damaged DNA (Chu and Chang, 1988) with a chemically defined footprint on both DNA strands (Reardon et al., 1993). The role of DDB1/2 in the pathway of excision repair is in initial recognition of a damaged site (Kapetanaki et al., 2006; Scrima et al., 2009; Robu et al., 2013) and subsequent activation of XPC/HR23B binding, and consequently suppresses UV mutagenesis (Tang et al., 2000). DDB2 expression is induced by UV light through p53 transactivation in human, but not mouse cells (Nichols et al., 2001; Tang and Chu, 2002). This provides a partial explanation for observations that excision repair of cyclobutane dimers is low in some mouse cell strains.
XPF (ERCC4)
The XP-F complementation group is rare, and the majority of cases have been found in Japan. Although the majority of patients have a mild photosensitive phenotype, mouse Xpf strains are very severely affected (Tian et al., 2004), suggesting that many mutations in XPF are inconsistent with viability and only mild mutations permit human survival and development (Niedernhofer et al., 2006). XPF is located on chromosome 16p13.3, and encodes a structure-specific endonuclease of 916 amino acids, which in association with the ERCC1 protein (de Laat et al., 1998), incises DNA on the 5' side of the damaged site to permit replacement synthesis to occur from the resulting 3’OH terminus (Sijbers et al., 1996, 1998; Bessho et al., 1997; Niedernhofer et al., 2004; Fagbemi et al., 2011).
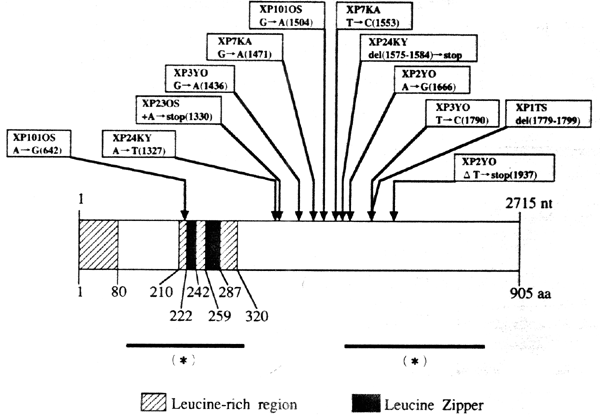
Figure 7. Location of the mutations in the XPF gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Reproduced from Matsumura et al., Human Molecular Genetics 7:969-974, 1998, with permission from Oxford University Press.]
XPG (ERCC5)
XPG is located on chromosome 13q32-33, and encodes an 1186 amino acid nuclease, which incises DNA 3' to the damaged site (Nouspikel and Clarkson, 1994; O'Donovan et al., 1994). XPG is a latent endonuclease that carries out the incision in vivo only after repair synthesis has proceeded from the initial cleavage by XPF/ERCC1 (Fagbemi et al., 2011). The gene is also known as ERCC5. The XP-G complementation group is rare, and most cases are known from Europe. Patients are severely affected and often exhibit combined symptoms of XP and CS (Nouspikel et al., 1997). These combined symptoms have been used to reveal a second function for XPG in the repair of oxidative damage (Nouspikel et al., 1997).
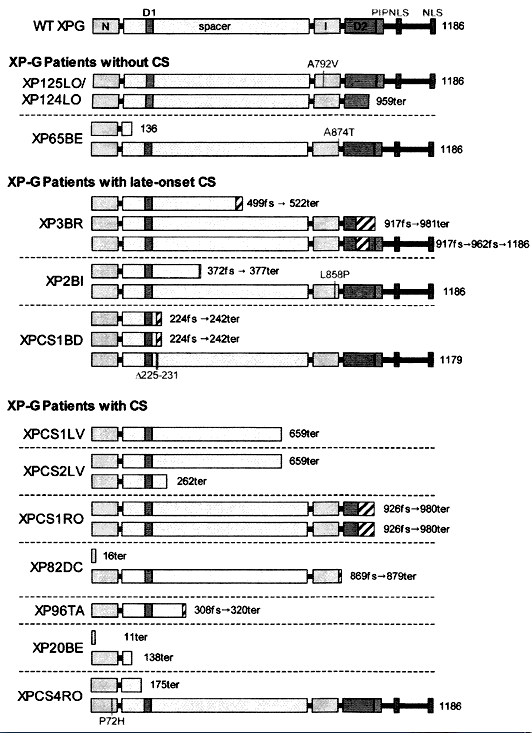
Figure 8. Location of the mutations in the XPG gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Reproduced from Scharer, XPG: Its products and biological roles, in Molecular mechanisms of xeroderma pigmentosum, Chapter 9, 2008, with permission from Landes Bioscience.]
XP-Variant
XP-V patients present with a similar clinical spectrum to those of XP-C patients, but were found to have normal excision repair but abnormal replication of damaged DNA (Cleaver, 1972; Lehmann et al., 1975). XPV is located on chromosome 6p21, and encodes a protein of 713 amino acids (Johnson et al. 1999; Masutani et al., 1999). The protein is a low-fidelity class Y DNA polymerase, variously known as hRad30A, pol η (eta) or Pol H (Ohmori et al., 2001). The complete human genomic sequence spans about 40kb, containing 10 coding exons and a cDNA of 2.14kb; exon I is untranslated and is 6kb upstream from the first coding exon (Thakur et al., 2001; Cleaver et al., 2003). Pol H can replicate UV-induced pyrimidine dimers in vivo with the insertion of the correct bases in the daughter strand; in vitro it is very error-prone and inserts mutagenic bases at about 1% frequency (Johnson et al., 2000; Matsuda et al., 2000; Ohashi et al., 2000).
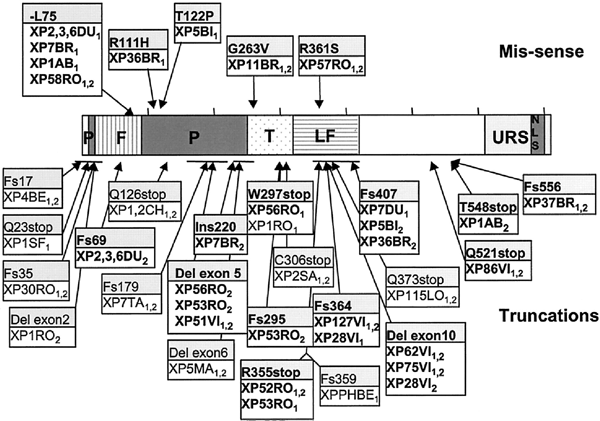
Figure 9. Location of the mutations in the XPV gene. This illustration is typical of a large number of mutations in this gene, but not necessarily all that have been reported. [Reproduced from Figure 4 of Broughton et al., in Proc Natl Acad Sci USA 99:815-820, 202 with permission.]
CSA (ERCC8)
CSA is located on chromosome 5, and encodes a 396 amino acid WD-repeat protein that is involved in the coupling between transcription and repair (Henning et al., 1995). The gene is also known as ERCC8. Cells with mutations in CSA fail to ubiquitinate RNA polymerase II after UV exposure (Bregman et al., 1996), and cannot remove and degrade the transcription complex stalled at a damaged site in DNA. The arrest of Pol II at damaged sites has been suggested as the most specific damage recognition process (Lindsey-Boltz and Sancar, 2007). The predominance of neurological and developmental disorders in CS patients suggests that CSA and CSB play a role in the repair of endogenous DNA damage, probably oxidative reactions in neural tissue (Cleaver and Revet, 2008; Frosina, 2008).
Several lines of evidence support the idea that the inadequate repair of endogenous damage may be the cause of CS neurodegeneration (see below for CSB). The capacity for oxidative damage repair is reduced in CSA cells, but to a lesser extent than in CSB (D'Errico et al., 2007).
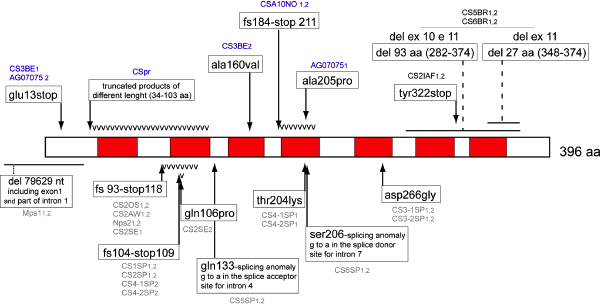
Figure 10. Location of the mutations in the CSA gene. Linear map of the mutations in the CSA/ERCC8 gene. Missense mutations are represented above the protein; other point mutations and deletions are represented beneath the protein. WD-repeat domains are shown in gray. [Reproduced from Laugel et al. (2008a) with permission of John Wiley and Sons.]
CSB (ERCC6)
CSB is located on chromosome 10q11-21, and encodes a 1493 amino acid protein with helicase motifs, and is also involved in the coupling between repair and transcription. The gene is also known as ERCC6. Cells with mutations in CSB also fail to ubiquitinate RNA polymerase II, and cannot remove and degrade the transcription complex stalled at a damaged site in DNA (Bregman et al., 1996). The protein has a nucleotide binding site, and acts as a DNA-dependent ATPase, but despite the helicase motifs at the sequence level, the protein does not appear to possess helicase activity in vitro (Selby and Sancar, 1997a, b; Citterio et al., 1998). Mutations in CSB can give rise to a wide range of symptoms ranging from mild UV sensitivity (Horibata et al., 2004) to the severe neonatal lethal disorder COFS (Laugel et al., 2008b), but a specific correlation between the site of mutation and the clinical severity has not yet been made.
A transposon, PGBD3 (“PiggyBac”-like) has been identified within intron 5 of the CSB gene, which functions as an alternative 39nt terminal exon (Newman et al., 2008). The alternatively spliced mRNA encodes an abundant chimeric protein in which CSB exons 2–5 are joined in frame to the PiggyBac transposase in a variety of human cell lines (exon 1 is untranslated). This chimeric protein continues to be expressed in primary CS cells, in which functional CSB is lost due to mutations beyond exon 5. The CSB-transposase fusion protein has been highly conserved for at least 43 Myr, since the divergence of humans and marmoset, but is not found in rodents. The suggestion has been made that this protein may cause CS in the absence of functional CSB protein, but this is unlikely for several reasons. CSB mutations are found both 3’ and 5’ to the intron 5 transposon (Figure 11), and mice with the Csb or Csa homolog knocked out give Cockayne-like disorders despite the absence of the transposon in rodents. The presence of the fusion protein is also not correlated with severity of CSB symptoms (Laugel et al., 2008a).
The predominance of neurological and developmental disorders in CS patients suggests that CSB plays a role in repair of endogenous oxidative DNA damage, originating most probably in mitochondria (Cleaver and Revet, 2008; Frosina, 2008). CSB mutant cells are sensitive to a variety of oxidants, and are unable to react to hypoxic stimuli by properly activating the hypoxia-inducible factor-1 (HIF-1) pathway (Filippi et al., 2008). CSB expression is under the control of HIF-1, and redistributes p300 between HIF-1 and p53 during hypoxic response (Filippi et al., 2008).
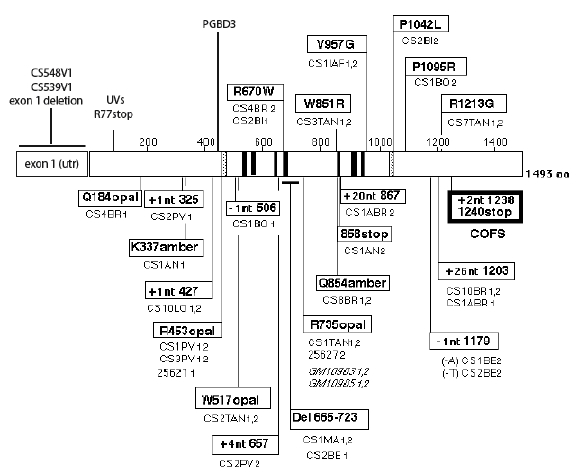
Figure 11. Location of the mutations in the CSB gene. Linear map of the mutations in the CSB/ERCC6 gene. Missense mutations are represented above the protein; other point mutations and deletions are represented beneath the protein. Helicase motifs (I to VI), acidic domain (A), and nuclear localization signal (N) are shown in gray. [Reproduced from Laugel et al. (2008a) with permission from John Wiley and Sons.]
Mitochondrial Deficiencies in Cockayne Syndrome
CSA and CSB have recently been found to play a role in mitochondrial functions, independent of their nuclear TCR functions, including repair of mitochondrial DNA damage, mitochondrial autophagy and the electron transport chain (Aamann et al., 2010; Kamenisch et al., 2010; Pascucci et al., 2012; Scheibye-Knudsen et al., 2012; Sykora et al., 2012). Mutations in CSA and CSB consequently influence the redox balance and mediate hypersensitivity to oxidative agents (Pascucci et al., 2012). Mitochondrial content is increased in CSBm/m mouse cells due to reduced autophagy, but the residual mitochondria are highly abnormal with increased free radical production (Scheibye-Knudsen et al., 2012). The failure of autophagy has been linked to neurodegeneration (Komatsu et al., 2006). The observation of specific abnormalities in the Purkinje cells of Csbm/m mice (Laposa et al., 2007) may be related to this mitochondrial function. The accumulation of damaged mitochondria in Purkinje cells can block the numerous branching extensions that establish up to 150,000 synaptic neuronal connections for each Purkinje cell (Girard et al., 2012) leading to Purkinje cell degeneration (Laposa et al., 2007). A role for CSA and CSB in mitochondrial functions puts Cockayne syndrome into context with many other human neurodegenerative diseases that have abnormal mitochondrial functions, some caused by mutations in mitochondrial DNA (Lax et al., 2011; Lenzken et al., 2011).
Mitochondria are deficient in the nucleotide excision repair of both cyclobutane dimers and [6-4] photoproducts (Clayton et al., 1974; Pascucci et al., 1997), but competent in base excision repair (Stevsner et al., 2008; Sykora et al., 2012). CSB appears to stimulate the activity of nuclear and mitochondrial 8-oxo-G glycosylase, but not thymine or uracil glycosylases, or single strand break repair, and interacts with PARP-1 and AP endonuclease (Stevsner et al., 2008). The accumulation of single base damage in vivo would be expected if the repair of 8-oxo-dG glycosylase was defective, but such evidence is at present limited (Wang et al., 2012).
Mitochondrial leakage of byproducts of oxidative phosphorylation is frequently invoked as a source of oxidative damage to DNA, especially in the context of neurodegeneration and autophagy in CS (Hoeijmakers, 2001; Barnes and Lindahl, 2004; Wallace, 2005; Koren and Kimchi, 2012; Scheibye-Knudsen et al., 2012), but there is little direct evidence. The cellular content of antioxidant enzymes, such as catalase and superoxide dismutase, and numerous cytoplasmic targets for oxidants that can absorb these byproducts, make it arguable that oxidants could reach the nucleus. There is also ample evidence for the formation of oxidative DNA adducts from a variety of other sources (Brooks, 2007; Kirkali et al., 2009) (Wang et al., 2012). The inflammatory (innate immune) response or oxidative burst from macrophages or neutrophils in peripheral blood, involving enzymes such as p450 oxidase and NADPH oxidase generates toxic oxidants. Glial cells in the brain that originate in the myeloid lineage, and which are increased in CS brain tissue, could similarly be a source of oxidative damage (Dedon and Tannenbaum, 2004; Graeber, 2010; Petersen et al., 2012). Nitric oxide, a neurotransmitter, has been shown to damage mitochondrial DNA (Druzhyna et al., 2005). Overstimulation of neuronal NMDA-type glutamate receptors activated NADPH oxidase (neuronal NOX2) and generated superoxide that damaged adjacent cells and induced apoptosis (Reyes et al., 2012). Glutamate also stimulated the induction of apurinic endonuclease 1, a major repair enzyme of oxidative DNA damage (Yang et al., 2010).
Additional Mutations Associated with UV Sensitive Patients
Some mildly UV-sensitive patients that do not have the typical developmental or neurological characteristics of CS patients, have provided cell lines that are UV sensitive and show normal global excision repair, but reduced transcription coupled repair, and a failure of RNA synthesis to recover from UV damage (Fujiwara et al., 1981; Cleaver et al., 1992). These have been defined as the UVs syndrome (Fujiwara et al. 1981). Some of these represent unusual manifestations of mutations in the known CSA (Nardo et al., 2009) and CSB genes (Horibata et al., 2004), but others appear to genuinely different (Itoh et al., 1994, 1995). In a survey of 106 clinically diagnosed XP patients, 6 could not be associated with mutations in any of the known XP genes A through G, or V, and may represent mutations in undiscovered genes (Bradford et al., 2011).
One case of UVs was found to have a termination mutation in the early region of the CSB gene, resulting in an apparent absence of the CSB protein (Horibata et al., 2004). Two other bona fide CSB patients have been reported, however, with a 5' deletion of the noncoding exon I and upstream sequences in CSB, and a similar complete lack of protein (Laugel et al., 2008a). These cases show extremely severe CS symptoms, inconsistent with the prediction from the UVs case (Horibata et al., 2004). The apparent lack of protein in the one UVs case with a CSB mutation cannot, therefore, be the sole cause of the mild symptoms. One possible explanation is that certain sequence contexts permit a small amount of gene translation through termination codons, producing low levels of protein undetectable by western blots. Some UVs cases are sensitive to UV radiation, but not to oxidative damage (Spivak and Hanawalt, 2006), suggesting that low levels of protein may be sufficient for the repair of oxidative damage, but not UV damage.
Mutations in a gene, UVSSA (also designated KIAA1530), have recently been identified as another cause of the UVs syndrome that, together with CSA and CSB, is a cofactor of RNA Pol II (see Table 2) (Cleaver, 2012; Nakazawa et al., 2012; Schwertman et al., 2012; Zhang et al., 2012). These coordinate a deubiquitin pathway that removes RNA Pol II from transcription blocking lesions (Anindya et al., 2010; Cleaver, 2012). CSA recruits UVSSA after UV damage to form a complex with USP7 (ubiquitin specific protease 7) that removes ubiquitin, stabilizes the ERCC6/RNA Pol II-complex, and restores the hypophosphorylated form of RNA Pol II (Fei and Che, 2012). Removal of the arrested RNA Pol II from the damaged site permits the excision of the damage by the later steps of the NER process, followed by resumption of transcription. The association of CSB with transcription elongation is also stabilized by DNA damage (Boom et al., 2004). RNA Pol II stalled on the DNA template is displaced as a result of deubiquitylation and the transcription-stalling damage is removed, allowing transcription to resume. Excision of the stalling lesion involves the later stages in the NER mechanism.
All reported mutations in CSA, CSB and UVSSA result in UV sensitivity and reduced TCR, including those associated with the mild UVs syndrome. This means that the reduced repair of UV damage in transcriptionally active genes is only consistently correlated with skin photosensitivity, but not with cancer or developmental and neurological disorders. The cause of the major developmental and neurological disorders in CS is more likely associated with other functions of CSA and CSB such as the oxidative stress response and mitochondrial autophagy that is defective in CS cells, but not necessarily in UVs cells (Spivak and Hanawalt, 2006; Pascucci et al., 2012).
Patient Care and Treatment
Clinical care for the repair deficient diseases has concentrated on prevention and treatment of skin cancers (Lambert et al., 2006). The neurological and other symptoms present a much greater challenge (Cleaver and Revet, 2008). For all the photosensitive diseases the paramount approach is early diagnosis and stringent protection from sun exposure (Lambert et al., 2006; Cleaver, 2008). While this approach is difficult and is not perfect, it does reduce the rate of onset of skin symptoms including cancers. The development of skin cancers can be treated with a variety of approaches, including 5-fluorouracil and imiquimod creams (Lambert et al., 2006). Retinoids have been used with some success, but there can be unacceptable side effects on bone strength (Kraemer et al., 1992; Lambert et al., 2006). Surgical excision of tumors may also be necessary, but due to the high frequency with which new tumors arise, considerable disfigurement may result, and metastatic disease may eventually occur.
Several gene therapy approaches have been attempted with XP. Yarosh and colleagues developed a DNA repair cream that delivers bacterial repair genes to the skin with some considerable success (Yarosh et al., 2001). Delivering corrected genes has also shown success in model systems, but in patients the difficulty would be in treating the whole area of sun-exposed skin (Arnaudeau-Bégard et al., 2003; Armelini et al., 2005; Marchetto et al., 2005).
Since a common belief attributes neurodegeneration to cellular damage from reactive oxygen, treatment with antioxidants could be one approach (Heidrick et al., 1984; Holloszy, 1998; Meydani et al., 1998). Treatment of Atm mice, that are deficient in double-strand break responses, with antioxidants has shown promising success in reducing Purkinje cell damage and improving behavioral endpoints (Chen et al., 2003; Gueven et al., 2006), and this could be attempted in Cs and Xp mice. A complication in using knockout mice, however, is that many NER-deficient mice show milder neurodegenerative symptoms than humans, and the cancer burden is higher, especially for Cs (van der Horst et al., 1997, 2002; Laposa et al., 2007) and Xpa (Berg et al., 1997; Tanaka et al., 2001).
A major regulatory problem regarding clinical trials in human repair deficient patients is that there are few patients with whom to design safety and effectiveness trials. One possibility would be to test drugs and antioxidants that have already been approved by FDA for use in Alzheimers, Parkinson’s, and other more common neurological disorders.
Several support groups are now available for patients with XP and CS that provide information, activities and practical help. For XP, these include The XP Society, and Family Support in the USA, and a Support Group in England. For CS, there is the Share and Care Network.
References
Aamann, M.D., Sorensen, M.M., Hvitby, C., Berquist, B.R., Muftuoglu, M., Tian, J., de Souza-Pinto, N.C., Scheibye-Knudsen, M., Wilson, D.M.r., Stevnsner, T., Bohr, V.A. (2010) Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB 24, 2334-2346.
Anindya, R., Mari, P.O., Kristensen, U., Kool, H., Giglia-Mari, G., Mullenders, L.H., Fousteri, M., Vermeulen, W., Egly, J.M., Svejstrup, J.Q. (2010) A ubiquitin-binding domain in Cockayne syndrome B required for transcription-coupled nucleotide excision repair. Mol Cell 38, 637-648.
Armelini, M.G., Muotri, A.R., Marchetto, M.C., de Lima-Bessa, K.M., Sarasin, A., Menck, C.F. (2005) Restoring DNA repair capacity of cells from three distinct diseases by XPD gene-recombinant adenovirus. Cancer Gene Ther 12, 389-396.
Arnaudeau-Bégard C, Brellier F, Chevallier-Lagente O, Hoeijmakers J, Bernerd F, Sarasin A, T., M. (2003) Genetic correction of DNA repair-deficient/cancer prone xeroderma pigmentosum group C keratinocytes. Hum Gene Ther 14, 983-986.
Asahina, H., Kuraoka, I., Shirakawa, M., Morita, E.H., Miura, N., Miyamoto, I., Ohtsuka, E., Okada, Y., Tanaka, K. (1994) The XPA protein is a zinc metalloprotein with an ability to recognize various kinds of DNA damage. Mutat Res 315, 229-237.
Barnes, D.E., Lindahl, T. (2004) Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Ann Rev Genet 445-476.
Berg, R.J.W., Vries, A.d., Steeg, H.v., Gruijl, F.R.d. (1997) Relative susceptibilities of XPA knockout mice and their heterozygous and wild-type littermates to UVB-induced skin cancer. Cancer Res 57, 581-584.
Bessho, T., Sancar, A., Thompson, L.H., Thelen, M.P. (1997) Reconstitution of human excision nuclease with recombinant XPF-ERCC1 complex. J Biol Chem 272, 3833-3837.
Boom, V.v.d., Citterio, E., Hoogstrraten, D., Zotter, A., Egly, J.-M., Cappellen, V.A.v., Hoeijmakers, H.J.H., Hooutsmuller, A.B., Vermeulen, W. (2004) DNA damage stabilizes interaction of CSB with the transcription elongation machinery. J Cell Biol 166, 27-36.
Bootsma, D., Kraemer, K.H., Cleaver, J.E., Hoeijmakers, J.H.J. (1998) Nucleotide excision repair syndromes: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. In: Vogelstein B., Kinzler K.W. (eds) The Genetic Basis of Human Cancer. McGraw-Hill, pp. 245-274.
Botta, E., Nardo, T., Broughton, B.C., Marinoni, S., Lehmann, A.R., Stefanini, M. (1998) Analysis of mutations in the XPD gene in Italian patients with trichothiodystrophy: site of mutation correlates with repair deficiency, but gene dosage appears to determine clinical severity. Amer J Hum Gen 63, 1036-1048
Botta, E., Offman, J., Nardo, T., Ricotti, R., Zambruno, G., Sansone, D., Balestri, P., Raams, A., Kleijer, W.J., Jaspers, N.G., Sarasin, A., Lehmann, A.R., Stefanini, M. (2007) Mutations in the C7orf11 (TTDN1) gene in six nonphotosensitive trichothiodystrophy patients: no obvious genotype-phenotype relationships. Hum Mutat 28, 92-96.
Bradford, P.T., Goldstein, A.M., Tamura, D.T., Khan, S.G., Ueda, T., Boyle, J., Oh, K.-S., Imoto, K., Inui, H., Moriwaki, S.-I., Emmert, S., Pike, K.M., Raziuddin, A., Plona, T.M., DiGiovanna, J.J., Tucker, M.A., Kraemer, K.H. (2011) Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet 48, 168-176.
Bregman, D.B., Halaban, R., van Gool, A.J., Henning, K.A., Friedberg, E.C., Warren.S.L. (1996) UV-induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci USA 93, 11586-11590.
Brooks, P.J. (2007) The case for 8,5'-cyclopurine-2'-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neurosci 145, 1407-1417.
Broughton, B.C., Lehmann, A.R., Harcourt, S.A., Arlett, C.F., Sarasin, A., Kleijer, W.J., Beemer, F.A., Nairn, R., Mitchell, D.L. (1990) Relationship between pyrimidine dimers, 6-4 photoproducts, repair synthesis and cell survival: studies using cells from patients with trichothiodystrophy. Mutat Res 235, 33-40.
Cadet, J., Vigney, P. (1990) The photochemistry of nucleic acids. In: Morrison H. (ed) Bioorganic photochemistry:photochemistry and the nucleic acids. John Wiley and Sons, New York, pp. 1-273.
Chen, P., Peng, C., Luff, J., Spring, K., Watters, D., Bottle, S., Furuya, S., Lavin, M.F. (2003) Oxidative stress is responsible for deficient survival and dendritogenesis in purkinje neurons from ataxia-telangiectasia mutated mutant mice. J Neurosci Res 23, 11453-11460.
Chiganças, V., Lima-Bessa, K.M., Stary, A., Menck, C.F., Sarasin, A. (2008) Defective transcription/repair factor IIH recruitment to specific UV lesions in trichothiodystrophy syndrome. Cancer Res 68, 6074-6083.
Chu, G., Chang, E. (1988) Xeroderma pigmentosum group E cells lack a nuclear factor that binds to damaged DNA. Science 242, 564-567.
Citterio, E., Rademakers, S., van der Horst, G.T., van Gool, A.J., Hoeijmakers, J.H.J., Vermeulen, W. (1998) Biochemical and biological characterization of wild-type and ATPase- deficient Cockayne syndrome B repair protein. J Biol Chem 273, 11844-11851.
Clayton, D.A., Doda, J.N., Friedberg, E.C. (1974) The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc Natl Acad Sci USA 71, 2777-2781.
Cleaver, J.E. (1968) Defective repair replication in xeroderma pigmentosum. Nature 218, 652-656
Cleaver, J.E. (1969) Xeroderma pigmentosum: a human disease in which an initial stage of DNA repair is defective. Proc Natl Acad Sci USA 63, 428-435
Cleaver, J.E. (1972) Xeroderma pigmentosum: variants with normal DNA repair and normal sensitivity to ultraviolet light. J Invest Dermatol 58, 124-128.
Cleaver, J.E. (1984) DNA repair deficiencies and cellular senescence are unrelated in xeroderma pigmentosum cell lines. Mech Aging Devel 27, 189-196.
Cleaver, J.E. (2004) Nucleotide excision repair and human disease. Encyclopedia of Biol Chem 3, 123-129.
Cleaver, J.E. (2005) Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nature Reviews on Cancer 5, 564-573.
Cleaver, J.E. (2008) Diagnosis of xeroderma pigmentosum and related DNA repair-deficient cutaneous diseases. Dermatol 13, 41-48.
Cleaver, J.E. (2012) Photosensitivity syndrome brings to light a new transcription-coupled DNA repair factor. Nature Genetics 44, 477-478.
Cleaver, J.E., Brennan-Minnella, A.M., Swanson, R. A., Fong, K. W., Chen, J., Chou, K.M., Chen, Y.W., Revet, I., Bezrookove, V. (2014) Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc Natl Acad Sci USA 111, 13487-13492.
Cleaver, J.E., Charles, W.C., McDowell, M., Karentz, D., Thomas, G.H. (1992) Are eight xeroderma pigmentosum groups (A-G,V) and two Cockayne syndrome groups (A,B) the whole story in DNA repair? In: Bohr V.A., Wasserman K., Kraemer K.H. (eds) DNA repair mechanisms. Vol 35: Alfred Benzon Symposium. Munksgaard, Copenhagen, pp. 56-67.
Cleaver, J.E., Charles, W.C., Thomas, G.H., McDowell, M.L. (1995) A deletion and an insertion in the alleles for the xeroderma pigmentosum (XPA) DNA-binding protein in mildly affected patients. Human Mol Gen 4, 1685-1687.
Cleaver, J.E., Collins, C., Ellis, J., Volik, S. (2003) Genome sequence and splice site analysis of low fidelity DNA polymerases H & I involved in replication of damaged DNA. Genomics 82, 561-570.
Cleaver, J.E., Crowley, E. (2002) UV damage, DNA repair and skin carcinogenesis. Frontiers in Bioscience 7, 1024-1043.
Cleaver, J.E., Revet, I. (2008) Clinical implications of the basic defects in Cockayne syndrome and xeroderma pigmentosum and the DNA lesions responsible for cancer, neurodegeneration and aging. Mech Ageing Dev 129, 492-497.
Cleaver, J.E., Thompson, L.H., Richardson, A.S., States, J.C. (1999) A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat 14, 9-22.
Coin, F., Bergmann, E., Tremeau-Bravard, A., Egly, J.-M. (1999) Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J 18, 1357-1366.
Coin, F., Marinoni, J.C., Rodolfo, C., Fribourg, S., Pedrini, A.M.a., Egly, J.M. (1998) Mutations in the XPD helicase gene result in XP and TTD phenotypes, preventing interaction between XPD and the p44 subunit of TFIIH. Nat Gen 20, 184-188.
Coin, F., Oksenych, V., Egly, J.M. (2007) Distinct roles for the XPB/p52 and XPD/p44 subcomplexes of TFIIH in damaged DNA opening during nucleotide excision repair. Mol Cell 26, 245-256.
Cooper, P.K., Nouspikel, T., Clarkson, S.G., Leadon, S.A. (1997) Defective transcription coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science 275, 990-993.
Courdavault, S., Baudouin, C., Charveron, M., Favier, A., Cadet, J., Douki, T. (2004) Larger yield of cyclobutane dimers than 8-oxo-7,8-dihydroguanine in the DNA of UVA-irradiated human skin cells. Mutat Res 556(1-2), 135-142.
Couve-Privat, S., Bouadjar, B., Avril, M.F., Sarasin, A., Daya-Grosjean, L. (2002) Significantly High Levels of Ultraviolet-specific Mutations in the Smoothened Gene in Basal Cell Carcinomas from DNA Repair-deficient Xeroderma Pigmentosum Patients. Can Res 62, 7185-7189.
D'Errico, M., Parlanti, E., Teson, M., Degan, P., Lemma, T., Calcagnile, A., Iavarone, I., Jaruga, P., Ropolo, M., Pedrini, A.M., Orioli, D., Frosina, G., Zambruno, G., Dizdaroglu, M., Stefanini, M., Dogliotti, E. (2007) The role of CSA in the response to oxidative DNA damage in human cells. Oncogene 26, 4336-4343.
de Boer, J., de Wit, J., van Steeg, H., Berg, R.J., Morreau, H., Visser, P., Lehmann, A.R., Duran, M., Hoeijmakers, J.H.J., Weeda, G. (1998a) A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Molec Cell 1, 981-990.
de Boer, J., Donker, I., de Wit, J., Hoeijmakers, J.H.J., Weeda, G. (1998b) Disruption of the mouse xeroderma pigmentosum group D DNA repair/basal transcription gene results in preimplantation lethality. Cancer Res 58, 89-94.
de Laat, W.L., Sijbers, A.M., Odijk, H., Jaspers, N.G., Hoeijmakers.J.H. (1998) Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res 26, 4146-4152.
Dedon, P.C., Tannenbaum, S.R. (2004) Reactive nitrogen species in the chemical biology of inflammation. Arch Biochem Biophys 423, 12-22.
Dixon, K.M., Deo, S.S., Wong, G., Slater, M., Norman, A.W., Bishop, J.E., Posner, G.H., Ishizuka, S., Halliday, G.M., Reeve, V.E., Mason, R.S. (2005) Skin cancer prevention: a possible role of 1,25dihydroxyvitamin D3 and its analogs. J Steroid Biochem Mol Biol 97, 137-143.
Driggers, W.J., Grishko, V.I., LeDoux, S.P., Wilson, G.L. (1996) Defective repair of oxidative damage in the mitochondrial DNA of a xeroderma pigmentosum group A cell line. Cancer Res 56, 1262-1266.
Druzhyna, N.M., Musiyenko, S.I., Wilson, G.L., LeDoux, S.P. (2005) Cytokines induce nitric oxide-mediated mtDNA damage and apoptosis in oligodendrocytes. Protective role of targeting 8-oxoguanine glycosylase to mitochondria. J Biol Chem 280, 21673-21679.
Dualan, R., Brody, T., Keeney, S., Nichols, A.F., Admon, A., Linn, S. (1995) Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics 29, 62-69.
Ellison, M.J., Childs, J.D. (1981) Pyrimidine CPDs induced in Escherichia coli DNA by ultraviolet radiation present in sunlight. Photochem Photobiol 34, 465-469.
Evans, E., Moggs, J.G., Hwang, J.R., Egly, J.-M., Wood, R.M. (1997) Mechanism of open complex formation by human nucleotide excision repair factors. The EMBO Journal 16, 6559-6573.
Fassihi, H., Sethi,M., Fawcett, H., Wing, J., Chandler, N., Mohammed, S.,Craythorne, E., Morley, A. M., Lim, R., Turner, S., Henshaw, T., Garrood, I., Giunti, P., Hedderly, T., Abiona, A., Naik, H., Harrop, G., McGibbon, D., Jaspers,N. G., Botta, E., Nardo, T., Stefanini, M., Young, A. R., Sarkany, R. P., Lehmann, A. R.. (2016) Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci USA 113, 1236-1245.
Fagbemi, A.F., Orelli, B., Scharer, O.D. (2011) Regulation of endonuclease activity in human nucleotide excision repair. DNA Repair 10, 722-729.
Faghri, S., Tamura, D., Kraemer, K.H., Digiovanna, J.J. (2008) Trichothiodystrophy: A systematic review of 112 published cases characterizes a wide spectrum of clinical manifestations. J Med Genet 45, 609-621.
Fei, J., Che, J. (2012) KIAA1530 protein is recruited by Cockayne syndrome complementation group protein A (CSA) to participate in transcription-coupled repair (TCR). J Biol Chem 287, 35118-35126.
Filippi, S., Latini, P., Frontini, M., Palitti, F., Egly, J.-M., Proietti De Santis, L. (2008) CSB is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. EMBO J 27, 2545-2556.
Frosina, G. (2008) Oxidatively damaged DNA repair defect in cockayne syndrome and its complementation by heterologous repair proteins. Curr Med Chem 15, 940-953.
Fujiwara, Y., Ichihashi, M., Kano, Y., Goto, K., Shimuzu, K. (1981) A new human photosensitive subject with a defect in the recovery of DNA synthesis after ultraviolet-light irradiation. J of Invest Derm 77, 256-263.
Garland, C., Garland, F. (1980) Do sunlight and vitamin D reduce the likelihood of colon cancer? Int J Epidemiol 9, 227-231.
Giglia-Mari, G., Coin, F., Ranish, J.A., Hoogstraten, D., Theil, A., Wijgers, N., Jaspers, N.G., Raams, A., Argentini, M., van der Spek, P.J., Botta, E., Stefanini, M., Egly, J.M., Aebersold, R., Hoeijmakers, J.H., Vermeulen, W. (2004) A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group Am. Nat Genet 36, 714-719.
Girard, M., Larivière, R., Parfitt, D.A., Deane, E.C., Gaudet, R., Nossova, N., Blondeau, F., Prenosil, G., Vermeulen, E.G., Duchen, M.R., Richter, A., Shoubridge, E.A., Gehring, K., McKinney, R.A., Brais, B., Chapple, J.P., McPherson, P.S. (2012) Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc Natl Acad Sci USA 109, 1661-1666.
Graeber, M.B. (2010) Changing face of microglia. Science 330, 783-788.
Graham, J.M.J., Anyane-Yeboa, K., Raams, A., Appeldoorn, E., Kleijer, W.J., Garritsen, V.H., Busch, D., Edersheim, T., G. , Jaspers, N.G. (2001) Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Amer J Hum Gen 69, 291-300.
Groisman, R., Polanowska, J., Kuraoka, I., Sawada, J.-I., Saijo, M., Drapkin, R., Kisselev, A.F., Tanaka, K., Nakatani, Y. (2003) The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell 113, 357-367.
Gueven, N., Luff, J., Peng, C., Hosokawa, K., Bottle.S., Lavin, M.F. (2006) Dramatic extension of tumor latency and correction of neurobehavioral phenotype in Atm-mutant mice with a nitroxide antioxidant. Free Radic Biol Med 41, 992-1000.
Hanawalt, P.C. (1994) Transcription-coupled repair and human disease. Science,266,1957-1958 He, Z., Henrickson, L.A., Wold, M.S., Ingles, C.J. (1995) RPA involvement in the damage-recognition and incision steps of nucleotide excision repair. Nature 374, 566-569.
Heidrick, M.L., Hendricks, L.C., Cook, D.E. (1984) Effect of dietary 2-mercaptoethanol on the life span, immune system, tumor incidence and lipid peroxidation damage in spleen lymphocytes of aging BC3F1 mice. Mech Ageing Dev 27, 341-358.
Henning, K.A., Li, L., Iyer, N., McDaniel, D., Reagan, M.S., Legerski, R., Schultz, R.A., Stefanini, M., Lehmann, A.R., Mayne, L.V., Friedberg, E.C. (1995) The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with the CSB protein and a subunit of RNA polymerase II, TFIIH. Cell 82, 555-564.
Hoeijmakers, J.H. (2001) Genome maintenance mechanisms for preventing cancer. Nature 411, 366-374.
Hoeijmakers, J.H.J., Egly, J.M., Vermeulen, W. (1996) TFIIH: a key component in multiple DNA transactions. Current Opin Genet Dev 6, 26-33.
Holloszy, J.O. (1998) Longevity of exercising male rats: effect of an antioxidant supplemented diet. Mech Ageing Dev 100, 211-219.
Horibata, K., Iwamoto, Y., Kuraoka, I., Jaspers, N.G., Kurimasa, A., Oshimura, M., Ichihashi, M., Tanaka, K. (2004) Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc Natl Acad Sci USA 101, 15410-15415.
Horn, S., Figl, A., Rachakonda, P.S., Fischer, C., Sucker, A., Gast, A., Kadel, S., Moll, I., Nagore, E., Hemminki, K., Schadendorf, D., Kumar, R. (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339, 959-961.
Huang, F.W., Hodis, E., Xu, M.J., Kryukov, G.V., Chin, L., Garraway, L.A. (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957-959.
Ikegami, T., Kuraoka, I., Saijo, M., Kodo, N., Kyogoku, Y., Morikawa, K., Tanaka, K., Shirakawa, M. (1998) Solution structure of the DNA- and RPA-binding domain of the human repair factor XPA. Nat Struct Biol 5, 701-706.
Itin, P.H., Sarasin, A., Pittelkow, M.R. (2001) Trichothiodystrophy: update on the sulfur-deficient brittle hair syndromes. J Amer Acad Derm 44, 891-920.
Itoh, T., Fujiwara, Y., Ono, T., Yamaizumi, M. (1995) UVs syndrome, a new general category of photosensitive disorder with defective DNA repair, is distinct from xeroderma pigmentosum variant and rodent complementation group I. Amer J Hum Gen 56, 1267-1276.
Itoh, T., Linn, S., Ono, T., Yamaizumi, M. (2000) Reinvestigation of the classification of five cell strains of xeroderma pigmentosum group E with reclassification of three of them. J Invest Dermatol 114, 1022-1029.
Itoh, T., Nichols, A., Linn, S. (2001) Abnomal regulation of DBB2 gene expression in xeroderma pigmentosum group E strains. Oncogene 20, 7041-7050.
Itoh, T., Ono, T., Yamaizumi, M. (1994) A new UV-sensitive syndrome not belonging to xeroderma pigmentosum or Cockayne syndrome: siblings showing biochemical characteristics of Cockayne syndrome without typical clinical manifestations. Mutat Res 314, 233-248.
Jaspers, N.G., Raams, A., Silengo, M.C., Wijgers, N., Niedernhofer, L.J., Robinson, A.R., Giglia-Mari, G., Hoogstraten, D., Kleijer, W.J., Hoeijmakers, J.H., Vermeulen, W. (2007) First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Amer J Hum Genet 80, 457-466.
Johnson, R.E., Kondratick, C.M., Prakash, S., Prakash, L. (1999) hRAD30 mutations in the variant form of xeroderma pigmentosum. Science 264, 263-265.
Johnson, R.E., Washington, M.T., Prakash, S., Prakash, L. (2000) Fidelity of human DNA polymerase h. J Biol Chem 275, 7447-7450.
Jones, C.A., Huberman, E., Cunningham, M.L., Peak, M.J. (1987) Mutagenesis and cytotoxicity in human epithelial cells by far- and near-ultraviolet radiations: action spectra. Rad Res 110, 244-254.
Jones, C.J., Wood, R.D. (1993) Preferential binding of the xeroderma pigmentosum group A complementing protein to damaged DNA. Biochem 32, 12096-12116.
Kamenisch, Y., Fousteri, M., Knoch, J., von Thaler, A.K., Fehrenbacher, B., Kato, H., Becker, T., Dollé, M.E., Kuiper, R., Majora, M., Schaller, M., van der Horst, G.T., van Steeg, H., Röcken, M., Rapaport, D., Krutmann, J., Mullenders, L.H., Berneburg, M. (2010) Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med 207, 379-390.
Kapetanaki, M.G., Guerrero-Santoro, J., Bisi, D.C., Hsieh, C.L., Rapic-Otrin, V., Levine, A.S. (2006) The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA 103, 2588-2593.
Keeney, S., Chang, G.J., Linn, S. (1993) Characterization of a human DNA damage binding protein implicated in xeroderma pigmentosum E. J Biol Chem 268, 21293-21300.
Kirkali, G., de Souza-Pinto, N.C., Jaruga, P., Bohr, V.A., Dizdaroglu, M. (2009) Accumulation of (5'S)-8,5'-cyclo-2'-deoxyadenosine in organs of Cockayne syndrome complementation group B gene knockout mice. DNA Repair (Amst) 8, 274-278.
Kleijer, W.J., Laugel, V., Berneburg, M., Nardo, T., Fawcett, H., Gratchev, A., Jaspers, N.G., Sarasin, A., Stefanini, M., Lehmann, A.R. (2008) Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 7, 744-750.
Komatsu, M., Waguri, S., Chiba, T., Murata, S., Iwata, J., Tanida, I., Ueno, T., Koike, M., Uchiyama, Y., Kominami, E., Tanaka, K. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880-886.
Koonin, E.V. (1993) Escherichia coli dinG gene encodes a putative DNA helicase related to a group of eukaryotic helicases including Rad3 protein. Nucl Acid Res 21, 1497-1503.
Koren, I., Kimchi, A. (2012) Promoting tumorigenesis by suppressing autophagy. Science 338, 889-890. Kraemer, K.H., DiGiovanna, J.J., Peck, G.L. (1992) Chemoprevention of skin cancer in xeroderma pigmentosum. J Dermatol 19, 715-718.
Kraemer, K.H., Lee, M.M., Andrews, A.D., Lambert, W.C. (1994) The role of sunlight and DNA repair in melanoma and nonmelanoma skin cancer. The xeroderma pigmentosum paradigm. Arch Derm 130, 1018-1021.
Kraemer, K.H., Lee, M.M., Scotto, J. (1984) DNA repair protects against cutaneous and internal neoplasia:evidence from xeroderma pigmentosum. Carcinogenesis 5, 511-514.
Kraemer, K.H., Lee, M.M., Scotto, J. (1987) Xeroderma pigmentosum:cutaneous, ocular and neurological abnormalities in 830 published cases. Arch Dermatol 123, 241-250.
Kuraoka, I., Monta, E.H., Saijo, M., Matsuda, T., Monkawa, K., Shirakawa.M., Tanaka, K. (1996) Identification of a damaged-DNA binding domain of the XPA protein. Mutat Res 362, 87-95.
Lambert, W.C., Gagna, C.E., Centurion, S.A. (2006) Xeroderma Pigmentosum. In: Lebwohl M.G., Heymann W.R., Berth-Jones J.B., Coulson J. (eds) Treatment of Skin Disease: Comprehensive Therapeutic Strategies. Mosby/Elsevier, London, pp. 694-698.
Lange, S.S., Mitchell, D.L., Vasquez, K.M. (2008) High mobility group protein B1 enhances DNA repair and chromatin modification after DNA damage. Proc Natl Acad Sci USA 105, 10320-10325.
Laposa, R.R., Huang, E.J., Cleaver, J.E. (2007) Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci USA 104, 1389-1394.
Laugel, V., Dalloz, C., Durand, M., Sauvanaud, F., Kristensen, U., Vincent, M.C., Pasquier, L., et al. (2010) Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum Mutat 31, 113-126.
Laugel, V., Dalloz, C., Stary, A., Cormier-Daire, V., Desguerre, I., Renouil, M., Fourmaintraux, A., Velez-Cruz, R., Egly, J.M., Sarasin, A., Dollfus, H. (2008a) Deletion of 5' sequences of the CSB gene provides insight into the pathophysiology of Cockayne syndrome. Eur J Hum Genet 16, 320-327.
Laugel, V., Dalloz, C., Tobias, E.S., Tolmie, J.L., Martin-Coignard, D., Drouin-Garraud, V., Valayannopoulos, V., Sarasin, A., Dollfus, H. (2008b) COFS syndrome: three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J Med Genet 45, 564-571.
Lax, N.Z., Turnbull, D.M., Reeve, A.K. (2011) Mitochondrial mutations: newly discovered players in neuronal degeneration. Neuroscientist 17, 645-658.
Lehmann, A.R., Arlett, C.F., Broughton, B.C., Harcourt, S.A., Steingrimsdottir, H., Stefanini, M., Taylor, A.M.R., Natarajan, A.T., Green, S., others. (1988) Trichothiodystrophy, a human DNA repair disorder with heterogeneity in the cellular response to ultraviolet light. Cancer Res 48, 6090-6096.
Lehmann, A.R., Kirk-Bell, S., Arlett, C.A., Paterson, M.C., Lohman, P.H.M., de Weerd-Kastelein, E.A., Bootsma, D. (1975) Xeroderma pigmentosum cells with normal levels of excision repair have a defect on DNA synthesis after UV-irradiation. Proc Natl Acad Sci USA 72, 219-235.
Lenzken, S., Romeo, V., Zolezzi, F., Cordero, F., Lamorte, G., Bonanno, D., Biancolini, D., Cozzolino, M., Pesaresi, M.G., Maracchioni, A., Sanges, R., Achsel, T., Carrì, M.T., Calogero, R.A., Barabino, S.M. (2011) Mutant SOD1 and mitochondrial damage alter expression and splicing of genes controlling neuritogenesis in models of neurodegeneration. Hum Mutat 32, 168-182.
Li, L., Lu, X., Peterson, C.A., Legerski, R.J. (1995a) An interaction between the DNA repair factor XPA and replication protein A appears essential for nucleotide excision repair. Molec Cell Biol 15, 5396-5402.
Li, L., Peterson, C.A., Lu, X., Legerski, R.J. (1995b) Mutations in XPA that prevent association with ERCC1 are defective in nucleotide excision repair. Molec Cell Biol 15, 1993-1998.
Li, Y., huang, T.-T., Carlson, E.J., Melov, S., Ursell, P.C., Olsen, J.L., Noble, L.J., Yoshimura, M.P., Burger, C., Chan, P.K., Wallace, D.C., Epstein, C.J. (1995c) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genetics 11, 376-381.
Lindsey-Boltz, L.A., Sancar, A. (2007) RNA poymerase: the most specific damage recognition protein in cellular responses to DNA damage. Proc Nat Acad Sci USA 104, 13213-13214.
Maher, V. M., Dorney, D. J., Mendrake, A. L., Konze-Thomas, B., McCormick, J. J. (1979) DNA excision repair processes in human cells can eliminate the cytotoxic and mutagenic consequences of ultraviolet irradiation. Mutat Res 62, 311-323.
Maldonado, J.L., Fridlyand, J., Patel, H., Jain, A.N., Busam, K., Kageshita, T., Ono, T., Albertson, D., Pinkel, D., Bastian, B.C. (2003) Determinants of BRAF mutations in primary melanoma. J Natl Cancer Inst 95, 1878-1890.
Marchetto, M., Muotri, A., Burns, B., Friedberg, E., Menck, C. (2005) Gene transduction in skin cells: preventing cancer in xeroderma pigmentosum mice. Proc Natl Acad Sci USA 101, 17759-17764.
Masutani, C., Kusumoto, R., Yamada, A., Dohmae, N., Yokol, M., Yuasa, M., Araki, M., Iwa, S., Takio, K., Hanoaka, F. (1999) The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase h. Nature 399, 700-704
Masutani, C., Sugasawa, K., Yanagisawa, J., Sonoyama, T., Ui, M., Enomoto, T., Takio, K., Tanaka, K., Spek, P.J.v.d., Bootsma, D., HoeijmakersJ.H.J., Hanoaka, F. (1994) Purification and cloning of a nucleotide excision repair complex involving the xeroderma pigmentosum group C protein and a human homologue of yeast RAD23. EMBO J 13, 1831-1843.
Matsuda, T., Bebenek, K., Masutani, C., Hanoaka, F., Kunkel.T.A. (2000) Low fidelity DNA synthesis by human DNA polymerase eta. Nature 404, 1011-1013.
Meydani, M., Lipman, R.D., Han, S.N., Wu, D., Beharka, A., Martin, K.R., Bronson, R., Cao, G., Smith, D., Meydani, S.N. (1998) The effect of long-term dietary supplementation with antioxidants. Ann NY Acad Sci 854, 352-360.
Mitchell, D.L., Cleaver, J.E. (1990) Photochemical alterations of cytosine account for most biological effects after ultraviolet irradiation. Trends Photochem Photobiol 1, 107-119.
Mitchell, D.L., J.Jen, J.E.Cleaver (1991) Relative induction of cyclobutane dimers and cytosine photohydrates in DNA irradiated in vitro and in vivo with ultraviolet C and ultraviolet B light. Photochem Photobiol 54, 741-746.
Mitchell, D.L., J.Jen, J.E.Cleaver (1992) Sequence specificity of cyclobutane pyrimidine dimers in DNA treated with solar (ultraviolet B) radiation. Nucleic Acids Res 20, 225-229.
Mitra, D., Luo, X., Morgan, A., Wang, J., Hoang, M.P., Lo, J., Guerrero, C.R., Lennerz, J.K., Mihm, M.C., Wargo, J.A., Robinson, K.C., Devi, S.P., Vanover, J.C., D'Orazio, J.A., McMahon, M., Bosenberg, M.W., Haigis, K.M., Haber, D.A., Wang, Y., Fisher, D.E. (2012) An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 491, 449-453.
Miura, M., Miyamoto, I., Asahina, H., Satokata, I., Tanaka, K., Okada, Y. (1991) Identification and characterization of xpac protein, the gene product of the human XPAC (xeroderma pigmentosum group A complementing) gene. J Biol Chem 266, 19786-19789.
Miyamoto, I., Miura, N., Niwa, H., Miyazaki, J., Tanaka, K. (1992) Mutational analysis of the structure and function of the xeroderma pigmentosum group A complementing protein. Identification of essential domains for nuclear localization and DNA excision repair. J Biol Chem 267, 19786-19789.
Mouret, S., Baudouin, C., Charveron, M., Favier, A., Cadet, J., Douki, T. (2006) Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc Natl Acad Sci USA 103, 13765-13770.
Mu, D., Tursun, M., Duckett, D.R., Drummond, J.T., Modrich, P., Sancar, A. (1997) Recognition and repair of compound DNA lesions (Base damage and mismatch) by human mismatch repair and excision repair systems. Molec Cell Biol 17, 760-769.
Nakazawa, Y., Sasaki, K., Mitsutake, N., Matsuse, M., Shimada, M., Nardo, T., Takahashi, Y., Ohyama, K., Ito, K., Mishima, H., Nomura, M., Kinoshita, A., Ono, S., Takenaka, K., Masuyama, R., Kudo, T., Slor, H., Utani, A., Tateishi, S., Yamashita, S., Stefanini, M., Lehmann, A.R., Yoshiura, K.-i., Ogi, T. (2012) KIAA1530/UVSSA is responsible for UV-sensitive syndrome that facilitates damage-dependent processing of stalled RNA polymerase IIo in TC-NER. Nature Genetics 44, 586-592.
Nance, M.A., Berry, S.A. (1992) Cockayne syndrome: review of 140 cases. Amer J Med Genet 42, 68-84.
Nardo, T., Oneda, R., Spivak, G., Vaz, B., Mortier, L., Thomas, P., Orioli, D., Laugel, V., Stary, A., Hanawalt, P.C., Sarasin, A., Stefanini, M. (2009) A UV-sensitive syndrome patient with a specific CSA mutation reveals separable roles for CSA in response to UV and oxidative DNA damage. Proc Natl Acad Sci USA 106, 6209-6214.
Newman, J.C., Bailey, A.D., Fan, H.Y., Pavelitz, T., Weiner, A.M. (2008) An abundant evolutionarily conserved CSB-PiggyBac fusion protein expressed in Cockayne syndrome. PLoS Genet 4, e1000031.
Nichols, A.F., itoh, T., Graham, J.A., Lui, W., Yamaizumi, M., Linn, S. (2001) Human damage-specific DNA binding protein p48. Characterization of XPE mutations and regulation following UV irradiation. J Biol Chem 275, 21422-21428.
Nichols, A.F., Ong, P., Linn, S. (1996) Mutations specific to the xeroderma pigmentosum group E Ddb- phenotype. J Biol Chem 271, 24317-24320.
Niedernhofer, L.J., Garinis, G.A., Raams, A., Lalai, A.S., Robinson, A.R., Appeldoorn, E., Odijk, H., Oostendorp, R., Ahmad, A., van Leeuwen, W., Theil, A.F., Vermeulen, W., van der Horst, G.T., Meinecke, P., Kleijer, W.J., Vijg, J., Jaspers, N.G., Hoeijmakers, J.H. (2006) A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038-1043.
Niedernhofer, L.J., Odijk, H., Budzowska, M., van Drunen, E., Maas, A., Theil, A.F., de Wit, J., Jaspers, N.G., Beverloo, H.B., Hoeijmakers, J.H., Kanaar, R. (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand cross-link-induced double-strand breaks. Mol Cell Biol 24, 5776-5787.
Niggli, H.J., Cerutti, P.A. (1983) Cyclobutane-type pyrimidine photodimer formation and excision in human skin fibroblasts after irradiation with 313-nm ultraviolet light. Biochem 22, 1390-1395.
Nishigori, C., Moriwaki, S., Takebe, H., Tanaka, T., Imamura, S. (1994) Gene alterations and clinical characteristics of xeroderma pigmentosum group A patients in Japan. Arch Derm 130, 191-197.
Nishigori, C., Zghal, M., Yagi, T., Imamura, S., Komoun, M.R., Takebe, H. (1993) High prevalence of point mutations in exon 6 of xeroderma pigmentosum groupA-complementing (XPAC) gene in xeroderma pigmentosum group A patients in Tunisia. Amer J Hum Gen 53, 1001-1006.
Nouspikel, T., Clarkson, S.G. (1994) Mutations that disable the DNA repair gene XPG in a xeroderma pigmentosum group G patient. Hum Molec Gen 3, 963-967.
Nouspikel, T., Lalle, P., Leadon, S.A., Cooper, P.K., Clarkson, S.G. (1997) A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc Natl Acad Sci (USA) 94, 3116-3121.
O'Donovan, A., Davies, A.A., Moggs, J.G., West, S.C., Wood, R.D. (1994) XPG endonuclease makes the 3' incision in human DNA nucleotide excision repair. Nature 371, 432-435.
Oh, K.S., Imoto, K., Boyle, J., Khan, S.G., Kraemer, K.H. (2007) Influence of XPB helicase on recruitment and redistribution of nucleotide excision repair proteins at sites of UV-induced DNA damage. DNA Repair (Amst) 6, 1359-1370.
Ohashi, E., Ogi, T., Kusumoto, R., Iwai, S., Hanoaka, F., Ohmori, H. (2000) Error-prone bypass of certain DNA lesions by the human DNA polymerase k. Genes and Devel 14, 1589-1594.
Ohmori, H., Friedberg, E.C., Fuchs, R.P.P., Goodman, M.F., Hanaoka, F., Hinkle, D., Kunkel, T.A., Lawrence, C.W., Livneh, Z., Nohmi, T., Prakash, L., Prakash, S., Todo, T., Walker, G.C., Wang, Z., Woodgate, R. (2001) The Y-Family of DNA Polymerases. Molec Cell 8, 7-8.
Park, C.H., Mu, D., Reardon, J.T., Sancar, A. (1995) The general transcription factor TFIIH is recruited to the excision repair complex by the XPA protein independent of the TFIIE transcription factor. J Biol Chem 270, 4896-4902.
Pascucci, B., Lemma, T., Iorio, E., Giovannini, S., Vaz, B., Iavarone, I., Calcagnile, A., Narciso, L., Degan, P., Podo, F., Roginskya, V., Janjic, B.M., Van Houten, B., Stefanini, M., Dogliotti, E., D'Errico, M. (2012) An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 11, 520-529.
Pascucci, B., Versteegh, A., van Hoffen, A., van Zeeland, A.A., Mullenders, L.H., Dogliotti, E. (1997) DNA repair of UV photoproducts and mutagenesis in human mitochondrial DNA. J Molec Biol 273, 417-427.
Petersen, A.J., Rimkus, S.A., Wassarman, D.A. (2012) ATM kinase inhibition in glial cells activates the innate immune response and causes neurodegeneration in Drosophila. Proc Natl Acad Sci USA 109, 4039-4040.
Pfeifer, G.P., Drouin, R., Riggs, A.D., Holmquist, G.P. (1991) In vivo mapping of a DNA adduct at nucleotide resolution: Detection of pyrimidine [6-4] pyrimidone photoproducts by ligation-mediated polymerase chain reaction. Proc Natl Acad Sci USA 88, 1374-1378.
Pfeifer, G.P., R.Drouin, A.D.Riggs, G.P.Holmquist (1992) Binding of transcription factors creates hotspots for UV photoproducts. Molec Cell Biol 12, 1798-1804.
Ranish, J.A., Hahn, S., Lu, Y., Yi, E.C., Li, X.J., Eng, J., Aebersold, R. (2004) Identification of TFB5, a new component of general transcription and DNA repair factor IIH. Nat Genet 36, 707-713.
Reardon, J.T., Nichols, A.F., Keeney, S., Smith, C.A., Taylor, J.S., Linn, S., Sancar, A. (1993) Comparative analysis of binding of human damaged DNA-binding protein (XPE) and Escherichia coli damage recognition binding protein (UvrA) to the major ultraviolet photoproducts: T[c,s]T, T[t,s]T, T[6-4]T, and T[Dewar]T. J Biol Chem 268, 21301-21308.
Reardon, J.T., Sancar, A. (2005) nucleotide excision repair. Progr Nucleic Acids Res Mol Biol 79, 183-235.
Reid-Bayless, K. S., Arron, S. T., Loeb, L. A., Bezrookove, V., Cleaver, J. E. (2016) Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc Natl Acad Sci USA 113, 10151-101516.
Reyes, R.C., Brennan, A.M., Shen, Y., Baldwin, Y., Swanson, R.A. (2012) Activation of Neuronal NMDA Receptors Induces Superoxide-Mediated Oxidative Stress in Neighboring Neurons and Astrocytes. J Neurosci 32, 12973-12978
Robins, P., Jones, C.J., Biggerstaff, M., Lindahl, T., Wood, R.D. (1991) Complementation of DNA repair in xeroderma pigmentosum group A extracts by a protein with an affinity for damaged DNA. EMBO J 10, 3913-3921.
Robu, M., Brind'Amour, J., Kandan-Kulangara, F., Petitlclerc, N., Shah, R.G., Shah, G. (2013) Role of poly(ADP-ribose) polymerase-1 in the removal ofUV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci USA 110, 1658-1663.
Satokata, I., Iwa, K., Matsuda, T., Okada, Y., Tanaka, K. (1993) Genomic characterization of the human DNA excision repair-controlling gene XPAC. Gene 136, 345-348.
Scheibye-Knudsen, M., Ramamoorthy, M., Sykora, P., Maynard, S., Lin, P.C., Minor, R.K., Wilson, D.M.r., Cooper, M., Spencer, R., de Cabo, R., Croteau, D.L., Bohr, V.A. (2012) Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med 209, 855-869.
Schwertman, P., Lagarou, A., Dekkers, D.H.W., Raams, A., van der Hoek, A.C., Laffeber, C., Hoeijmakers, J.H.J., Demmers, J.A.A., Fousteri, M., Vermeulen, W., Marteijn, J.A. (2012) UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nature Genetics 44, 598-602..
Scrima, A., Konícková, R., Czyzewski, B.K., Kawasaki, Y., Jeffrey, P.D., Groisman, R., Nakatani, Y., Iwai, S., Pavletich, N.P., Thomä, N.H. (2009) Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 135, 1213-1223.
Selby, C.P., Sancar, A. (1997a) Cockayne syndrome group B protein enhances elongation by RNA polymerase II. Proc Natl Acad Sci USA 94, 11205-11209.
Selby, C.P., Sancar, A. (1997b) Human transcription-repair coupling factor CSB/ERCC6 is a DNA- stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J Biol Chem 272, 1885-1890.
Sijbers, A.M., De Laat, W.L., Ariza, R.R., Biggerstaff, M., Wei, Y.-F., Moggs, J., Carter, K.C., Shell, B.K., Evans, E., De Jong, M.C., Rademakers, S., De Rooiji, J., Jaspers, N.G.J., Hoeijmakers, J.H.J., Wood, R.D. (1996) Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86, 811-822.
Sijbers, A.M., van Voorst Vader, P.C., Snoek, J.W., Raams, A., Jaspers, N.G., Kleijer, W.J. (1998) Homozygous R488W point mutation in the XPF gene of a patient with xeroderma pigmentosum and late-onset neurologic disease. J Invest Dermatol 110, 832-836.
Sollitto, R.B., Kraemer, K.H., DiGiovanna, J.J. (1997) Normal vitamin D levels can be maintained despite rigorous photoprotection: six years' experience with xeroderma pigmentosum. J Am Acad Dermatol 37, 942-947.
Spina, C.S., Tangpricha, V., Uskokovic, M., Adorinic, L., Maehr, H., Holick, M.F. (2006) Vitamin D and cancer. Anticancer Res 26, 2515-2524.
Spivak, G., Hanawalt, P.C. (2006) Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 5, 13-22.
States, J.C., McDuffie, E.R., Myrand, S.P., McDowell, M., Cleaver, J.E. (1998) Distribution of mutations in the human xeroderma pigmentosum group A gene and their relationships to the functional regions of the DNA damage recognition protein. Hum Mutat 12, 103-113.
Stefanini, M., Lagomarsini, P., Giliani, S., Nardo, T., Botta, E., Pesericio, A., Kleijer, W.J., Lehmann, A.R., Sarasin, A. (1993) Genetic heterogeneity of the excision repair defect associated with trichothiodystrophy. Carcinogenesis 14, 1101-1105.
Sterenborg, H.J.C.M., van der Leun, J.C. (1990) Tumorigenesis by a long wavelength UV-A source. Photochem Photobiol 51, 325-330.
Stevsner, T., Muftuoglu, M., Aamann, M.D., Bohr, V.A. (2008) The role of cockayne syndrome group B (CSB) protein in base excision repair and aging. Mech Ageing Dev 129, 441-448.
Sugasawa, K., Ng, J.M.Y., Masutani, C., Iwai, S., van der Spek, P.J., Eker, A.P.M., Hanoaka, F., Bootsma, D., Hoeijmakers, J.H.J. (1998) Xeroderma pigmentosum group C protein complex is the initiator of global nucleotide excision repair. Molec Cell 2, 223-232.
Sugasawa, K., Okuda, Y., Saijo, M., Nishi, R., Matsuda, N., Chu, G., Mori, T., Iwai, S., Tanaka, K., Tanaka, K., Hanaoka, F. (2005) UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell 121, 387-400.
Svoboda, D.L., Briley, L.P., Vos, J.-M.H. (1998) Defective bypass replication of a leading strand cyclobutane thymine dimer in xeroderma pigmentosum variant cell extracts. Cancer Res 58, 2445-2448.
Sykora, P., Wilson, D.M.r., Bohr, V.A. (2012) Repair of persistent strand breaks in the mitochondrial genome. Mech Ageing Dev 133, 169-175.
Takeda, N., Shibuya, M., Maru, Y. (1999) The BCR-ABL oncoprotein potentially interacts with the xeroderma pigmentosum B protein. Proc Natl Acad Sci USA 96, 203-207.
Tanaka, K., Kamiuchi, S., Ren, Y., Yonemasu, R., Ichikawa, M., Murai, H., Yoshino, M., Takeuchi, S., Saijo, M., Nakatsu, Y., Miyauchi-Hashimoto, H., Horio, T. (2001) UV-induced skin carcinogenesis in xeroderma pigmentosum group A (XPA) gene-knockout mice with nucleotide excision repair-deficiency. Mutat Res 477, 31-40.
Tang, J., Chu, G. (2002) Xeroderma pigmentosum complementation group E and UV-damaged DNA-binding protein. DNA Repair 1, 601-616.
Tang, J.Y., Hwang, B.J., Ford, J.M., Hanawalt, P.C., Chu, G. (2000) Xeroderma pigmentosum p48 gene enhances global genomic repair and suppresses UV-induced mutagenesis. Molec Cell 5, 737-744.
Taylor, E.M., Broughton, B.C., Botta, E., Stefanini, M., Sarasin, A., Jaspers, N.G., Fawcett, H., Harcourt, S.A., Arlett, C.F.a., Lehmann, A.R. (1997) Xeroderma pigmentosum and trichothiodystrophy are associated with different mutations in the XPD (ERCC2) repair/transcription gene. Proc Nat Acad Sci USA 94, 8658-8663.
Taylor, J.S., Cohrs, M.P. (1987) DNA, light. and Dewar pyrimidinones: the structure and significance of TpT3. J Amer Chem Soc 109, 2834-2835.
Thakur, M., Wernick, M., Collins, C., Limoli, C., Crowley, E., Cleaver, J.E. (2001) DNA polymerase h undergoes alternative splicing, protects against UV sensitivity and apoptosis, and suppresses Mre11-dependent recombination. Genes, Chromosomes and Cancer 32, 222-235.
Thompson, L.H. (1998) Nucleotide excision repair: Its relation to human disease. In: Nickoloff J.A., Hoekstra M. (eds) DNA Repair in Higher Eukaryotes. Vol 2:: DNA Damage and Repair. Humana Press, Totowa, NJ, pp. 335-393.
Tresini, M., Warmerdam, D. O., Kolovos, P., Snijder, L., Vrouwe, M. G., Demmers, J. A., van IJcken, W. F., Grosveld, F .G., Medema, R. H., Hoeijmakers, J. H., Mullenders, L. H., Vermeulen, W., Marteijn, J. A. (2015) The core spliceosome as target and effector of non-canonical ATM signalling. Nature 523, 53-58.
Tian, M., Shinkura, R., Shinkura, N., Alt, F.W. (2004) Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol Cell Biol 24, 1200-1205.
Topping, R.S., Myrand, S.P., Williams, B.L., Albert, J.C., States, J.C. (1995) Characterization of the human XPA promoter. Gene 166, 341-342.
Tyrrell, R.M., Keyse, S.M. (1990) New trends in photobiology: the interaction of UVA radiation with cultured cells. J Photochem Photobiol B 4, 349-361.
Tyrrell, R.M., Pidoux, M. (1986) Endogenous glutathione protects human skin fibroblasts against the cytotoxic action of UVB, UVA and near-visible radiations. Photochem Photobiol 44, 561-564.
Tyrrell, R.M., Pidoux, M. (1987) Action spectra for human skin cells: estimates of the relative cytotoxicity of the middle ultraviolet, near ultraviolet, and violet regions of sunlight on epidermal keratinocytes. Cancer Res 47, 1825-1829.
van der Horst, G.T., Meira, L., Gorgels, T.G., de Wit, J., Velasco-Miguel, S., Richardson, J.A., Kamp, Y., Vreeswijk, M.P., Smit, B., Bootsma, D., Hoeijmakers, J.H., Friedberg, E.C. (2002) UVB radiation-induced cancer predisposition in Cockayne syndrome group A (Csa) mutant mice. DNA Repair 1, 143-157.
van der Horst, G.T., van Steeg, H., Berg, R.J., van Gool, A.J., de Wit, J., Weeda, G., Morreau, H., Beems, R.B., van Kreijl, C.F., de Gruijl, F.R., Bootsma, D., Hoeijmakers, J.H. (1997) Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 89, 425-435.
van der Pluijm, I., Garinis, G.A., Brandt, R.M., Gorgels, T.G., Wijnhoven, S.W., Diderich, K.E., de Wit, J., Mitchell, J.R., van Oostrom, C., Beems, R., Niedernhofer, L.J., Velasco, S., Friedberg, E.C., Tanaka, K., van Steeg, H., Hoeijmakers, J.H., van der Horst, G.T. (2006) Impaired genome maintenance suppresses the growth hormone--insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biology 5, e2.
Wakasugi, M., Sancar, A. (1998) Assembly, subunit composition, and footprint of human DNA repair excision nuclease. Proc Natl Acad Sci USA 95, 6669-6674.
Wakasugi, M., Sancar, A. (1999) Order of assembly of human DNA repair excision nuclease. J Biol Chem 274, 18759-18768.
Wallace, D.C. (2005) A mitochondrial paradigm of metabolic and degenerative diseases, aging and cancer:a dawn for evolutionary medicine. Ann Rev Genetics 39, 359-407.
Wang, J., Cao, H., You, C., Yuan, B., Bahde, R., Gupta, S., Nishigori, C., Niedernhofer, L.J., Brooks, P.J., Wang, Y. (2012) Endogenous formation and repair of oxidatively induced G[8-5 m]T intrastrand cross-link lesion. Nucleic Acids Res 40, 7368-7374.
Wang, N.J., Sanborn, Z., Arnett, K.L., Bayston, L.J., Liao, W., Proby, C.M., Leigh, I.M., et al. (2011) Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc Natl Acad Sci USA 108, 17761–17766.
Wang, Q.E., Zhu, Q., Wani, G., Chen, J., Wani, A.A. (2004) UV radiation-induced XPC translocation within chromatin is mediated by damaged-DNA binding protein, DDB2. Carcinogenesis 25, 1033-1043.
Wang, Y., Digiovanna, J.J., Stern, J.B., Hornyak, T.J., Raffeld, M., Khan, S.G., Oh, K.S., Hollander, M.C., Dennis, P.A., Kraemer, K.H. (2009) Evidence of ultraviolet type mutations in xeroderma pigmentosum melanomas. Proc Natl Acad Sci USA 106, 6279-6284.
Weeda, G., Ham, R.C.A.v., Vermeulen, W., Bootsma, D., Eb, A.J.v.d., Hoeijmakers, J.H.J. (1990) A presumed helicase encoded by ERCC-3 is involved in the human repair disorders xeroderma pigmentosum and Cockayne's syndrome. Cell 62, 777-791.
Yang, J.L., Tadokoro, T., Keijzers, G., Mattson, M.P., Bohr, V.A. (2010) Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1. J Biol Chem 285, 28191-28198.
Yarosh, D., Klein, J., O'Connor, A., Hawk, J., Rafal, E., Wolf, P. (2001) Effect of topically applied T4 endonuclease V in liposomes on skin cancer in xeroderma pigmentosum: a randomised study. Xeroderma Pigmentosum Study Group. Lancet 357, 926-929.
Zhang, W.R., Garrett, G.L., Arron, S.T., and Cleaver, J.E. (2016) Survey of Cockayne patients reports no skin cancers despite DNA repair deficiency. J Amer Acad Dermatol 74, 1270-1272.
Zhang, X., Horibata, K., Saijo, M., Ishigami, C., Ukai, A., Kanno, S.-i., Tahara, H., Neilan, E.G., Honma, M., Nohmi, T., Yasui, A., Tanaka, K. (2012) Mutations in KIAA1530/UVSSA cause UV-sensitive syndrome destabilizing ERCC6 in transcription-coupled DNA repair. Nature Genetics 44, 593-597.
11/02/08
04/06/13
09/06/16