Adaptation of Rod Photoreceptors
to Light and Dark
Małgorzata Różanowska
Cardiff Centre for Vision Science, Cardiff University
Maindy Road, Cardiff CF24 4LU, Wales, United Kingdom
RozanowskaMB@cf.ac.uk
Introduction
The illuminance of the eye at the Earth's surface varies by nearly 11 orders of magnitude during the normal day-night cycle; yet we are able to see differences between different luminous intensities nearly over this entire range (Rodieck, 1998; Fain et al., 2001; Arshavsky and Burns, 2012). Several mechanisms have evolved to allow the eye to function over a wide range of illuminance levels, so we can see the world around us. One of them is the action of iris muscles, which change the pupil diameter, and therefore are responsible for varying illuminance levels reaching the retina in a range of about two orders of magnitude. Although adjusting the pupil size to the changes in ambient light is important, it can only partly explain the ability of human vision to adjust to the wide range of light levels.
Other important adaptive changes occur in photoreceptor cells in the retina, where the photons are absorbed, and first steps leading to visual perception take place. Upon the absorption of photons by the visual pigments in the photoreceptor outer segment, a cascade of events is triggered leading to the hyperpolarisation of the photoreceptor plasma membrane, spreading rapidly to its synaptic terminal, where it leads to an inhibition of neurotransmitter release (Figure 1).
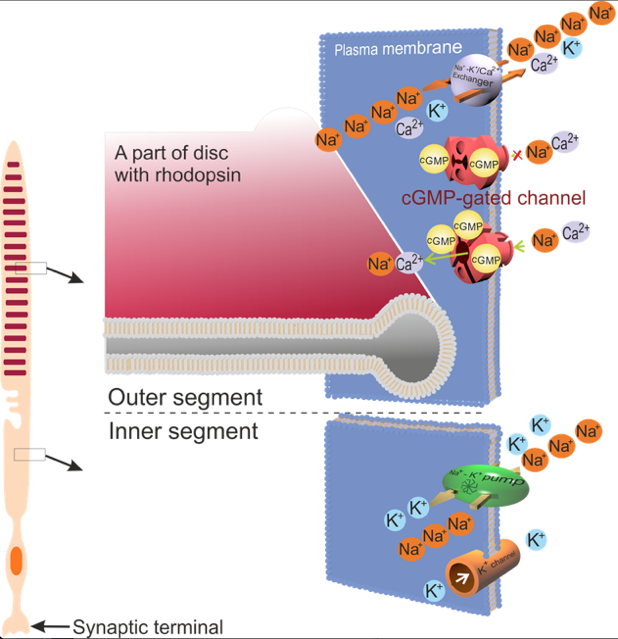
Figure 1. Schematic diagram of a rod and its cation channels in the inner and outer segments. In the dark, a fraction of the cGMP-gated channels remain open, which creates a current of Na+ circulating between the outer and inner segments: Na+ enters the outer segment via the cGMP-gated channel, and is removed by the Na+/K+ pump in the inner segment. Ca2+ entering the outer segment through the cGMP-gated channel are removed by the Na+/K+-Ca2+ exchanger present also in the outer segment. Absorption of a photon by the visual pigment, rhodopsin present in discs in the outer segment, initiates a cascade of events leading to the closure of the cGMP-gated channels in the photoreceptor outer segments. As a result, the absolute value of the circulating current of cations (mainly Na+) is decreased.
There are two types of photoreceptors in the retina: rods and cones. A dark-adapted rod can produce measurable responses upon absorption of a single photon; hyperpolarisation of its plasma membrane and a decrease in an absolute value of the circulating current of cations. An increase in the numbers of absorbed photons by rods leads to an increase in the response amplitudes until reaching a saturation level, where the plasma membrane is maximally hyperpolarised, and the circulating current of cations is close to zero (Figure 2; see also: “Retina: Photoreceptors and Functional Organization”). At the level of light where the saturation of the dark-adapted rod response is observed, the cones start producing measurable responses, and their responsiveness to flashes of light increase until reaching saturation at light intensities about 10,000 times greater than those producing just detectable responses in rods. Altogether, when dark-adapted and tested with short flashes of light, the rods and cones seem to cover only four orders of magnitude of illuminance (Kraft et al., 1993; Schneeweis and Schnapf, 1995).
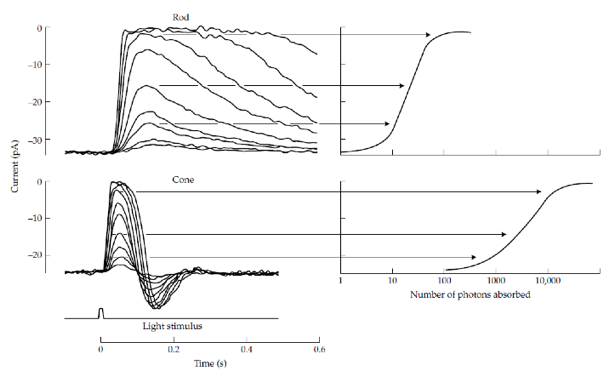
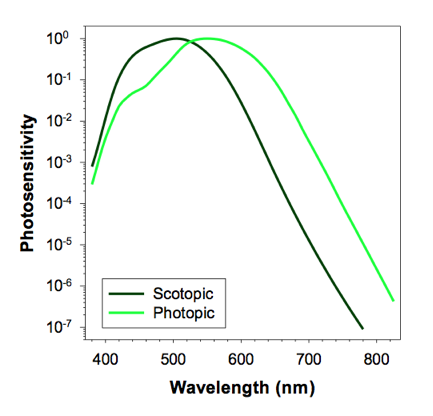
Figure 2. Top panels: Responses of the circulating currents of cations in dark-adapted rods and cones to flashes of light of different intensities (left). Right: the maximal amplitudes of the changes in the circulating currents as a function of the number of absorbed photons. (From: "Retina: Photoreceptors and Functional Organization") Bottom panel: Photosensitivity of the human eye under dim light, where vision is mediated by rods (scotopic conditions), and in bright light, where vision is mediated by cones (photopic conditions), normalized to the corresponding maxima.
Yet, when the photoreceptors are exposed to continuous ambient light, a flash of light that causes a saturated response in a photoreceptor adapted to the dark, can still elicit a response well below the saturation (Schneeweis and Schnapf, 1999; Schneeweis and Schnapf, 2000). This ability of photoreceptors to avoid saturation and respond to an increased flux of incident photons at different levels of ambient light is due to several changes occurring in these cells. The changes allow photoreceptors to adapt to different levels of ambient light by adjusting the sensitivity by adjusting the amplification factor of the phototransduction cascade, and the rate of recovery of cGMP levels. The amplification factor of the phototrasduction cascade can be controlled by: 1) the lifetime of rhodopsin in its biochemically active state, metarhodopsin II, and the availability of G protein, transducin in proximity to metarhodopsin II; 2) the lifetime of the activated transducin complex with phosphodiesterase 6 (PDE6); and 3) the affinity of the cGMP-gated channels for cGMP (Figure 3).
This chapter discusses these adaptive changes occurring in rod photoreceptors to maximize their sensitivity in the dark, as well as to maintain their responsiveness to increasing fluxes of absorbed light when exposed to ambient light. For more details, the reader is referred to several comprehensive reviews on the adaptation of photoreceptors to dark and light (Fain et al., 2001; Lamb and Pugh, 2004; Lamb and Pugh, 2006; Sampath and Fain, 2009; Fain, 2011; Arshavsky and Burns, 2012; Korenbrot, 2012a; Tang et al., 2013).
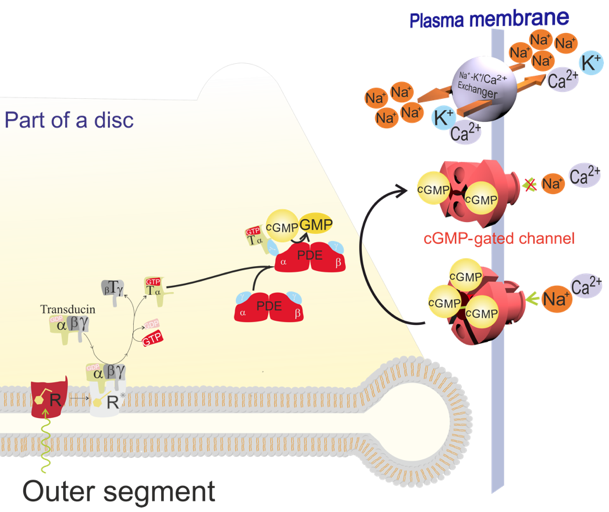
Figure 3. Diagram depicting essential steps in the phototransduction cascade in rods. Upon absorption of a photon by rhodopsin (R), the rhodopsin chromophore, 11-cis-retinal, is isomerized to all-trans-retinal. This is followed by a conformational conversion of rhodopsin to biochemically active metarhodopsin II (R*). Metarhodopsin II can bind a heterotrimeric G protein, transducin (T). The binding enables an exchange of the GDP nucleotide bound to the subunit α of transducin for a GTP, which is normally present in the cytoplasm. The binding of GTP activates transducin, which then dissociates from the metarhodopsin II as an activated α subunit with bound GTP (Tα-GTP) separated from the transducin subunits β and γ (Tβγ). Metarhodopsin II can then activate another transducin while the active Tα-GTP can bind one of two inhibitory subunits γ of the cGMP phosphodiesterase 6 (PDE). This relieves one of the two catalytic subunits of PDE6, α or β, from inhibition. The activated PDE hydrolyzes cGMP to GMP, and remains active as long as it binds to Tα-GTP. As a result of PDE action, the concentration of cGMP in the cytoplasm of the photoreceptor outer segment is reduced, which causes the cGMP to dissociate from the cGMP-gated channels. The cGMP-gated channels with fewer than three cGMP molecules bound to them close. The closure of the cGMP-gated channels reduces the influx of Na+ and Ca2+ into the photoreceptor outer segments. Because the Na+/K+ pumps in the inner segment pump Na+ out of the photoreceptors, the build-up of positive charge outside makes the photoreceptor plasma membrane hyperpolarized. The hyperpolarisation spreads to the synaptic terminal where it inhibits the release of glutamate. While the influx of Ca2+ through the cGMP-gated channels is disabled, the efflux of Ca2+ continues via the Na+/Ca2+-K+ exchanger. Thus, the closure of the cGMP-gated channels leads not only to a decrease in the cytoplasmic concentration of Na+, but also of Ca2+. The decrease in Ca2+ concentration acts as a signalling event partly responsible for the termination of phototransduction, restoring the initial state in the dark, as well as adaptation to background light.
High Sensitivity in Dark-Adapted Rods
In the dark-adapted rods, fluxes of dim light providing just a single photon incident on the rod outer segment can lead to the absorption of that single photon by rhodopsin, and can produce a measurable change in the circulating current (~1 pA) (Figure 2), and hyperpolarization of the plasma membrane, because the probability of photon absorption and amplification in phototransduction are the highest (Baylor et al., 1979).
The high probability of photon absorption is due to the high absorption coefficient of rhodopsin, which for the wavelength of 500 nm is 40,600 M-1cm-1, and dense packing of rhodopsin within discs, hundreds of which are stacked on top of one another within the rod outer segment (Wald and Brown, 1953). This provides a specific optical density for 500 nm light equal to 0.015 per µm of rod outer segment length (Rodieck, 1998). In the primate retina, rods have the longest outer segments in the parafovea, where their lengths are 35.2 µm. Thus, it can be estimated that 70% of incident photons of 500 nm wavelength will be absorbed by the dark-adapted parafoveal rods. In the periphery, the rod outer segments are the shortest with an approximate length of 23.9 µm, but still they enable absorption of 56% of incident photons. Rhodopsin has a high quantum yield of photoactivation; 67% of absorbed photons leads to an isomerisation of 11-cis-retinal followed by the formation of metarhodopsin II. Altogether, the probability of a 500 nm photon, incident on a dark-adapted rod, to be absorbed and activate rhodopsin is 47% and 38% in the primate parafovea and periphery, respectively.
The high amplification in the dark-adapted rods is possible because: 1) the lifetime of rhodopsin in its biochemically active state, metarhodopsin II, is the longest, and transducin concentration in proximity to metarhodopsin II is the highest, thus providing the opportunity to activate the maximal number of transducins; 2) the lifetime of activated transducin complexed with PDE6 is the longest, thus the complex can decompose the maximal number of cGMP molecules; and 3) the affinity of cGMP-gated channels for cGMP is the smallest, so the channels respond rapidly to the drop of the cytoplasmic concentration of cGMP. The mechanisms responsible for setting the maximal gains in these three amplification steps are discussed below.
The First Amplification Step: the activation of many transducins by a single metarhodopsin II. The activated rhodopsin, metarhodopsin II, can activate several transducin molecules, which are abundant in the outer segment of dark-adapted photoreceptors, accounting for a ~10-12% molar equivalent of rhodopsin (Fu and Yau, 2007; Arshavsky and Burns, 2012). The estimates of the rate of transducin activation by a single metarhodopsin II can range from 10 to over 3,000 per second at room temperature. In murine rods at physiological body temperature, a single metarhodopsin II can activate about 10-20 transducins within its lifetime of about 40-80 ms (Krispel et al., 2006; Burns and Pugh, 2009; Burns and Pugh, 2010; Chen et al., 2010a).
The short lifetime of metarhodopsin II is due to its rapid inactivation by rhodopsin kinase and arrestin, arrestin-1 (Figure 4) (Gross and Burns, 2010). The arrestin expressed in rods is arrestin-1. Rod outer segments contain two kinases, which can phosphorylate rhodopsin in vitro: G-protein-coupled receptor kinase 1 (GRK1), and protein kinase C (PKC). GRK1 is a single subunit membrane-associated protein, which phosphorylates only the activated state of rhodopsin, metarhodopsin II, whereas PKC phosphorylates with similar rates the dark-adapted and light-activated rhodopsin, as well as opsin without retinal (Greene et al., 1995; Greene et al., 1997; Williams et al., 1997; Maeda et al., 2003). In contrast to GRK1, whose activity is the lowest at high concentrations of free cytoplasmic Ca2+ observed in dark-adapted rods, and increases in response to a Ca2+ concentration ([Ca2+]) decrease, the activity of PKC is stimulated by Ca2+, and decreases when [Ca2+] decreases (Williams et al., 1997). The role of PKC in modulating the photoresponse has been studied in both dark-adapted and light-adapted salamander rods by comparing the photoresponses generated in the presence and absence of PKC activators and inhibitors (Xiong et al., 1997). A lack of significant effects of the PKC activators and inhibitors on photoresponses indicates that PKC does not play a significant role in the phototransduction modulation in amphibian rods, and/or its changed activity can be adequately counterbalanced by another kinase.
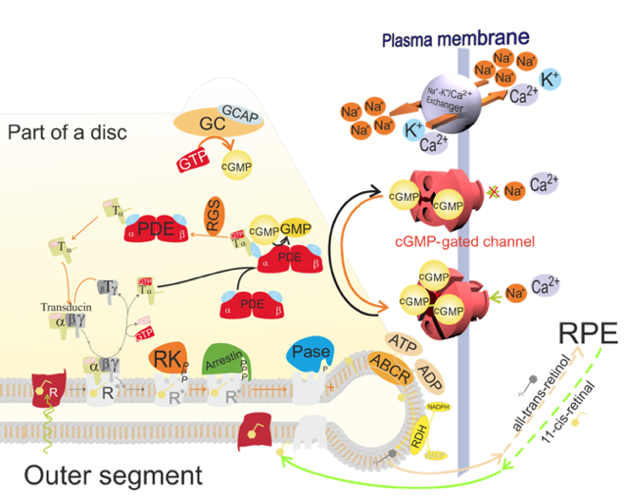
Figure 4. Activation (black arrows) and deactivation (orange arrows) steps in the phototransduction cascade. Activation steps are described in Figure 3. To terminate the phototrasduction, metarhodopsin II (R*) and the complex of phosphodiesterase 6 (PDE)-transducin (T)α with bound GTP need to be deactivated, and cGMP levels in the cytoplasm need to be restored. Metarhodopsin II is deactivated by the phosphorylation of its cytoplasmic terminal by rhodopsin kinase (RK), and the subsequent binding of arrestin, which further disables binding to transducin. The PDE-Tα(GTP) complex is deactivated upon hydrolysis of GTP to GDP in the α subunit of transducin, a reaction which is facilitated by the regulator of the G protein signalling (RGS) complex. This is followed by the dissociation of Tα(GDP), and the return of PDE to its inactive state. Subsequently, the Tα subunit reassociates with the Tβγ subunits to form an inactive heterotrimer.
While an increased number of cGMP-gated channels are closing, thus disabling an influx of Ca2+, the Na+/Ca2+-K+ exchanger continues to extrude Ca2+ from the outer segment. This leads to a decrease of intracellular Ca2+ concentration. The decreased concentration of Ca2+ acts as a signalling event partly responsible for the termination of the phototransduction cascade, restoring the state in the dark and the adaptation to background light. A low cytoplasmic Ca2+ concentration leads to the activation of RK and the activation of guanylate cyclase (GC). The latter process is mediated by the calcium-binding proteins, guanylate cyclase activating proteins (GCAPs). The activated GC synthesizes cGMP from GTP, leading to a recovery of the cGMP concentration. Moreover, a decrease in intracellular Ca2+ concentration leads to the dissociation of Ca2+ from calmodulin, and the subsequent dissociation of calmodulin from the cGMP-gated channel, which results in an increase in affinity of the cGMP-gated channel for cGMP. When more cGMP-gated channels reopen, the influx of Na+ and Ca2+ to the cytoplasm is increased, and the polarization of the plasma membrane changes towards its state in the dark.
On the other hand, knocking out Grk1 in mice results in a dramatic increase in the duration of photoresponses, indicating that GRK1 is the major contributor to the phosphorylation of rhodopsin in vivo, and it is essential for the normal deactivation of the phototransduction cascade (Chen et al., 1999). Depending on the species, the cytoplasmic carboxy terminus of rhodopsin has six to seven phosphorylation sites. Phosphorylation of at least two or preferably three serine/threonine residues enables metarhodopsin II to bind on its cytoplasmic surface to arrestin (Mendez et al., 2000a; Mendez et al., 2000b; Doan et al., 2006; Vishnivetskiy et al., 2007; Kim et al., 2013). Although the phosphorylation of metarhodopsin II partly hampers its ability to activate transducins, the binding of arrestin to phosphorylated metarhodopsin II abolishes this ability completely (Nikonov et al., 2008).
The essential role of rhodopsin phosphorylation in shortening the metarhodopsin II lifetime is underscored by experiments on transgenic mice expressing, as a fraction of total rhodopsin, a transgenic rhodopsin with a truncated carboxy terminus, so that the phosphorylation sites are absent (Chen et al., 1995). A fraction of the photoresponses recorded from the transgenic rods exhibit extended durations and increased amplitudes, similar to the photoresponses recorded in Rrk1-/- rods (Chen et al., 1999). The ratio of these abnormal photoresponses to all recorded responses corresponds to the ratio of transgenic rhodopsin to the total rhodopsin expressed in these mice (Chen et al., 1995).
It has been estimated that GRK1 accounts for only 1-2% of the molar equivalent of rhodopsin in wild-type rods, and therefore, it can be expected that the rate of phosphorylation of metarhodopsin II will be proportional to the concentration of active GRK1 (Chen et al., 2012). The activity of GRK1 can be regulated by recoverin (also known as S-modulin), which is a small 23 kDa protein belonging to a family of calcium-binding proteins (Makino et al., 2004; Chen et al., 2010a; Chen et al., 2012).
Recoverin consists of four EF-hand domains, two of which are active in binding Ca2+. The N-terminal glycine is post-translationally modified by myristoylate. The binding of calcium strongly affects the conformation of recoverin, and consequently its anchorage in the disc membrane and interaction with GRK1. In the dark-adapted state, characterized by a high cytoplasmic concentration of free calcium cations, two Ca2+ are bound to recoverin, and the myristoyl chain protrudes from the protein interior and anchors the recoverin into the disc membrane. This anchoring allows recoverin to maintain in the dark its highest concentration in proximity to GRK1, accounting for 0.6 mM recoverin being present in the outer segment. In contrast to Ca2+-free recoverin that does not bind to GRK1, the recoverin-Ca2+ complex binds to the N terminus helix of GRK1, which, in turn, prevents from conformational change of GRK1 needed for the phosphorylation of metarhodopsin II (Higgins et al., 2006; Komolov et al., 2009; Zernii et al., 2011).
There is growing body of evidence indicating that rhodopsin and metarhodopsin II form dimers and larger oligomers in the disc membrane (Fotiadis et al., 2003; Fotiadis et al., 2006; Knepp et al., 2012; Sommer et al., 2012; Jastrzebska et al., 2013). Based on EPR measurements, Myers and colleagues proposed that Ca2+ induces the dimerization of recoverin (Myers et al., 2013). The Ca2+-bound recoverin dimer binds to the N-terminal of GRK1, which prevents GRK1 from interacting with the metarhodopsin II dimer (Ames et al., 2006; Komolov et al., 2009).
Thus, it can be expected that in the dark-adapted rods, recoverin-Ca2+ complex can inhibit the phosphorylation of metarhodopsin II, thereby allowing for the longest lifetime of biochemically active metarhodopsin II. Extrapolating further, it can be expected that in the absence of recoverin-mediated inhibition, metarhodopsin II will be rapidly phosphorylated, so that the photoresponse will not reach the same amplitude as in the presence of recoverin, and will terminate faster. Interestingly, however, these predictions are only partly fulfilled in vivo. In the recoverin-deficient rods, the photoresponses do terminate faster than in the wild-type rods, but their amplitudes are surprisingly similar to the wild-type rods (Chen et al., 2012).
An overexpression of GRK1 can also be expected to result in an increased rate of metarhodopsin II phosphorylation, and consequently a faster termination of photoresponse, so its amplitude cannot build up as large as in the wild-type rods. Such an overexpression can be achieved by inserting an additional gene coding for GRK1 into the mouse genome, the expression of which is driven by the rod opsin promoter (Chen et al., 2012). This approach can provide transgenic mice with a 12-fold increased GRK1, in comparison to wild-type mice. Although the photoresponses in rods overexpressing GRK1 do terminate faster, their amplitudes exhibit only a 1.8-fold decrease. On the other hand, a deficiency of GRK1 leads to only a 1.7-fold increase in the photoresponse amplitude, which then persists over a period of several seconds (Chen et al., 1999). These observations suggest that a decreased or increased lifetime of active metarhodopsin II can be compensated by another mechanism(s), so the photoresponse amplitudes are maintained within a narrow range, even when the phosphorylation of metarhodopsin II occurs faster or slower. One of these mechanisms will be discussed below in the section on "Modulation of the photoresponse by cGMP synthesis".
In addition to the regulation by recoverin, the activity of GRK1 can also be regulated by the phosphorylation of its Ser21 residue by protein kinase A (PKA) (Osawa et al., 2011). The phosphorylated GRK1 exhibits a diminished ability to phosphorylate rhodopsin. In the mouse retina, the greatest concentration of phosphorylated GRK1 is observed in the dark-adapted animals. PKA is activated by high levels of cyclic AMP (cAMP). The main enzyme responsible for the synthesis of cAMP in photoreceptors is adenylyl cyclase type 1 (AC1). AC1 is activated by a Ca2+-calmodulin complex. Calmodulin is a ubiquitous Ca2+-binding protein, which is present at about a 1:700 ratio to rhodopsin, as determined in the bovine rod outer segments (Kohnken et al., 1981).
A surprising finding is that mice deficient in transducin α, and therefore unable to proceed with the phototransduction cascade leading to the decrease in the cytoplasmic free [Ca2+], still demonstrate dramatic changes in the phosphorylation level of GRK1, dependent on whether the mouse is adapted to dark or light. It has been shown in vitro on AC1-expressing HEK-293 cells that, once activated by intracellular Ca2+, AC1 activity can be further increased by activations of receptors coupled to a stimulatory G protein (Wayman et al., 1994). It is unclear whether or not a stimulatory G protein is involved in the activation of AC1 in photoreceptors, and, if so, which receptor activates that G protein, and what activates the receptor.
In vitro, the Ca2+-calmodulin complex can inhibit GRK1 directly by binding to a specific site on GRK1, which is different than the binding site for recoverin (Grigoriev et al., 2012). Calmodulin exerts its inhibitory effect on GRK1 at concentrations of calmodulin and free Ca2+, which are relevant for their physiological range in rods. An inhibition of GRK1 activity by 50% is induced by ~14 µM calmodulin saturated with Ca2+, whereas the concentration of recoverin required to induce a similar effect is only ~6 µM. The concentrations of calmodulin and recoverin in the rod outer segment have been estimated to be ~4 µM and ~600 µM, respectively (Kohnken et al., 1981; Reingruber et al., 2013). The concentration of Ca2+ needed for a 50% inhibition of GRK1 in vitro is ~1.4 µM for 30 µM recoverin, whereas only 0.4 µM for the same concentration of calmodulin (Grigoriev et al., 2012). These values are close to the physiological levels of [Ca2+] in the rod outer segment in the dark, where it is about 0.5 µM.
When both recoverin and calmodulin are present, the inhibitory effect increases in a synergistic way. In a 1:1 mixture of recoverin and calmodulin, the total concentration of both Ca2+-binding proteins needed for the 50% inhibition of GRK1 is only ~5 µM. It appears that the binding of one protein to GRK1 increases by ~50% the affinity of binding for the other protein. It has been proposed that under physiological conditions in the dark, both recoverin and calmodulin are bound to GRK1. This not only prevents the phosphorylation of metarhodopsin II, but also facilitates its efficient interaction with transducins undisturbed by GRK1, because the binding of the Ca2+-calmodulin complex prevents the GRK1 interaction with metarhodopsin II. When [Ca2+] decreases, Ca2+ dissociates first from recoverin, which subsequently dissociates from its binding site on GRK1 (Levay et al., 1998; Grigoriev et al., 2012). This decreases the GRK1 affinity for calmodulin, but still [Ca2+] needs to drop below 50 nM to cause the Ca2+ dissociation from calmodulin, and the subsequent dissociation of calmodulin from GRK1. Altogether, the mixture of calmodulin and recoverin seems to allow extending the modulatory effect of Ca2+ on GRK1 activity over a wider range of [Ca2+] than does a single protein.
Considering the effects of Ca2+-calmodulin complex on GRK1 activity, it is tempting to speculate that the relatively minor changes, observed in photoresponses of recoverin-knockout rods, in comparison with the wild-type rods, are due to a partial compensation for the absence of recoverin by an up-regulated expression of calmodulin. However, experimental results comparing the calmodulin expression in wild-type and recoverin-knockout rods have not yet been reported.
As discussed earlier, in the absence of GRK1, the lifetime of metarhodopsin II is considerably extended (Chen et al., 1999). Consequently, the time needed to reach the maximal amplitude in the Grk1-/- rods is longer than in Grk1+/+ rods. Also, the termination of the photoresponse in the knockout rods is considerably slowed down and follows a rather complex kinetics, in contrast to the wild-type rods, where the photoresponse termination follows an exponential decay. Clearly, in the Grk1-/- rods, the metarhodopsin II lifetime is the limiting factor in the rate of termination of phototransduction.
In addition to phosphorylation and inactivation by arrestin, there are two other pathways of metarhodopsin II deactivation: 1) a reversible transformation to an inactive state, metarhodopsin III; or 2) an irreversible hydrolysis of all-trans-retinal from opsin (Heck et al., 2003a; Zimmermann et al., 2004; Golobokova and Govardovskii, 2006; Gurevich and Gurevich, 2006; Sommer and Farrens, 2006; Sommer et al., 2006; Bartl and Vogel, 2007; Imai et al., 2007; Nymark et al., 2012). These pathways occur on much slower timescales than the inactivation by GRK1 and arrestin, and even slower than the deactivation of metarhodopsin II in the absence of GRK1. As already mentioned, in the mouse rods, the time constant for deactivation of metarhodopsin II by phosphorylation and capping by arrestin is less than 80 ms. In the absence of GRK1, the photoresponse terminates with a time constant of 3.3 s. On the other hand, the conversion of metarhodopsin II to metarhodopsin III has been reported to occur within 1-6 minutes, whereas the hydrolysis of the Schiff-base linkage of all-trans-retinal with opsin occurs with a time constant of ~5 minutes (Ebrey, 1968; Cone and Cobbs, 1969; Baumann and Bender, 1973; Bennett, 1980; Nymark et al., 2012). It has been demonstrated in a recent study on the isolated mouse retina that the decay of metarhodopsin II is biphasic, with the fast component concomitant with the formation metarhodopsin III occurring with a time constant of 3.6 min (Nymark et al., 2012). The decay of metarhodopsin III occurs with a time constant of 16 min, which is the same as the time constant of the slow component of metarhodopsin II decay.
The abundance of transducin in the dark-adapted outer segment facilitates its binding to metarhodopsin III. Upon binding with transducin, metarhodopsin III converts back to the biochemically most active state, metarhodopsin II, thus extending the time of phototransduction activation, which can further increase the amplification factor (Zimmermann et al., 2004).
The Second Amplification Step: the decomposition of many cGMP molecules by a single activated PDE6. A single transducin α subunit with bound GTP (Tα-GTP) can bind to a single PDE6 molecule, thus activating one of its catalytic subunits (Figures 3, 4). The second amplification step in the phototransduction cascade occurs via the degradation of several cGMP molecules by a single activated PDE6. It has been reported that the activated PDE6 hydrolyzes cGMP at a rate close to the limit set by aqueous diffusion (Leskov et al., 2000). In the mouse, the lifetime of the active transducin α-PDE6 complex is the longest in dark-adapted rods, where it lasts ~200 ms; therefore, the maximal number of cytoplasmic cGMP molecules can be decomposed by a single activated PDE6 before it becomes inactivated (Krispel et al., 2006; Chen et al., 2010a). This lifetime also corresponds to the time constant of the photoresponse duration typical for mammalian rods (Arshavsky and Burns, 2012). It also implies that the lifetime of the active transducin α-PDE6 complex can play an important role in setting the gain of the second amplification step in phototransduction. Eventually, PDE6 deactivation occurs when GTP in the transducin α subunit is hydrolyzed to GDP.
Transducin α itself exhibits a rather low GTPase activity. However, GTP hydrolysis is greatly accelerated upon binding with the regulator of the G protein signalling (RGS) protein complex, RGS9 (Pugh, 2006; Burns and Pugh, 2009; Kosloff et al., 2011). The RGS9 complex consists of three proteins: RGS9-1, Gβ5-L, and R9AP. RGS9-1 and Gβ5-L are essential for catalytic activity, whereas the transmembrane R9AP provides an anchor that binds the complex to the lipid membrane. Interestingly, to increase the GTPase activity, the RGS complex needs to interact also with the inhibitory subunit γ of PDE6. This interaction ensures that GTP hydrolysis occurs only after the activated α subunit of transducin binds to PDE6 γ. The hydrolysis of GTP to GDP in the subunit α of transducin causes a dissociation of the transducin α subunit, followed by return of PDE6 to an inactive state (Figure 4). It has been shown that the deactivation of the Tα-PDE6 complex is at least 2.5 times slower than the deactivation of metarhodopsin II by GRK1 and arrestin in the mouse rod, indicating that RGS activity is the major limiting factor for the rate of termination of the phototransduction cascade in the dark-adapted rods (Baker et al., 2006; Krispel et al., 2006).
It has been demonstrated on murine rods that all three components are essential for the activity of the RGS complex. In the absence of one of them, either RGS9-1, Gβ5-L, or R9AP, the time constant of termination of phototransduction elicited by a single photon is extended from 0.2 s in the wild-type rods to 2.0-2.6 s in the knockout rods (Chen et al., 2000; Krispel et al., 2003; Keresztes et al., 2004). Thus, an overexpression of the RGS9 complex in mouse rods, which can be achieved by an overexpression of R9AP, results in an accelerated termination of thephotoresponse (Krispel et al., 2006).
In the dark-adapted photoreceptors, most of the RGS9-1 and Gβ5-L are distributed outside the photoreceptor outer segments, suggesting the lowest RGS activity, and therefore the longest lifetime of the active Tα-PDE6 complex (Tian et al., 2013) (Figure 5). RGS9-1 is heavily phosphorylated at Ser475 in the dark or very dim light (below 1 lx) (Hu et al., 2001; Sokal et al., 2003; Tian et al., 2013). It has been shown in vitro that phosphorylation significantly reduces the RGS9-1 affinity for R9AP. Therefore, it has been suggested that the translocation of phosphorylated RGS9-1 out of the outer segment is due to its dissociation from R9AP, and a subsequent equilibration of its soluble form throughout the whole rod cell.
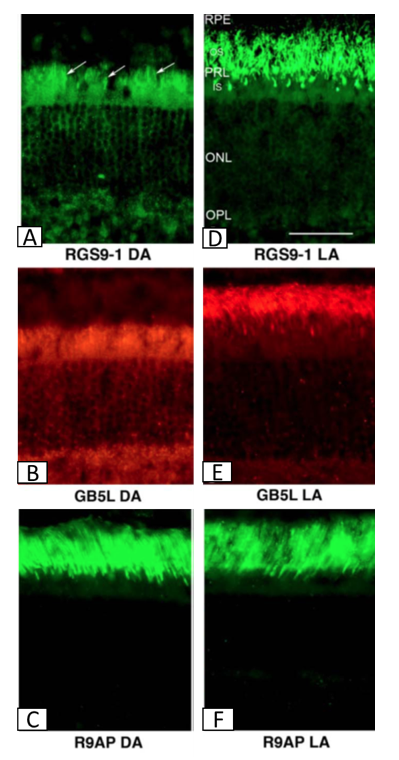
Figure 5. Distribution of the components of the RGS complex: RGS9-1, Gβ5-L, and R9AP in the dark-adapted (DA: A-C) and light-adapted (LA: D-F) mouse retina visualized by fluorescent microscopy. In the dark adapted retina, where mice were kept in dark for over 8 hours, RGS9-1 and Gβ5-L, the components responsible for the catalytic activity of the RGS complex, are distributed throughout the photoreceptor cells, mainly the inner segments (IS), cell bodies in the outer nuclear layer (ONL), and synaptic terminals in the outer plexiform layer (OPL) with very little, if any, being present in the outer segments (OS). Upon exposure to 200 lx of light, RGS9-1 and Gβ5-L translocate to the OS, so that eventually all three components are present in the OS layer in the light-adapted retina. The component anchored to the disc membrane, R9AP remains in the OS layer independent of the light/dark adaptation state. (Modified from Tian et al., 2013)
It has been suggested that the phosphorylation of RGS9-1 is due to protein kinase C (PKC) (Tian et al., 2013). The highest level of phosphorylation of RGS9-1 in dark-adapted photoreceptors is consistent with >85% of PKC activity being increased by high [Ca2+] (Balasubramanian et al., 2001; Hu et al., 2001; Sokal et al., 2003; Tian et al., 2013). PKC can phosphorylate not only RGS9-1, but also a protein phosphatase 2A (PP2A). The phosphorylation decreases PP2A activity, which otherwise could dephosphorylate RGS9-1. This is also consistent with observations of highly phosphorylated RGS9-1 in the dark, but not after exposure to light (Kirchhefer et al., 2014). However, as mentioned earlier, photoresponses of amphibian rods generated in the presence of PKC activators and inhibitors are almost identical to the photoresponses generated in their absence, thus questioning the substantial roles of PKC in phosphorylation of not only rhodopsin, but also RGS9-1 (Xiong et al., 1997). Because PKA activity is the highest in high [Ca2+], and decreases when free Ca2+ is depleted, it is seems worth considering whether or not PKA can contribute to RGS9-1 phosphorylation, and to compensate for the diminished PKC activity. Altogether, the role of PKC in mammalian rods, and the exact mechanism responsible for the phosphorylation of RGS9-1 in mammalian rods still needs to be elucidated.
To account for the accelerated rate of termination of phototransduction in rods depleted of recoverin or overexpressing GRK1, it has been suggested that the activated PDE6 lifetime can be regulated by recoverin and GRK1, but the mechanism responsible for that regulation is still unclear (Makino et al., 2004; Chen et al., 2012). It has been suggested that, once relieved from an inhibitory effect of recoverin, GRK1 may phosphorylate one of the proteins of the RGS complex, thus increasing its GTPase activity, and/or phosphorylate directly PDE6 γ or the transducin α subunit (Chen et al., 2012). The latter suggestion is supported by the structure of GRK1, where its N-terminal contains an RGS-like domain; however, it has not been tested directly yet if that domain can interact with transducin α and shorten the lifetime of its activated state (Siderovski et al., 1996). The potential role of the phosphorylation of PDE6 γ in modulating the lifetime of the active PDE6-transducin α complex is discussed below (second paragraph of “Effect of exposure to light on the second step in amplification cascade”).
If indeed GRK1 can regulate the lifetime of the activated PDE6-transducin α complex, the longest lifetime in the dark is due to an inhibitory effect of recoverin on GRK1 activity, which otherwise could increase the GTP-ase activity of the Tα(GTP)-PDE6 complex by the phosphorylation of an unknown target protein. According to that model, the recoverin knockout rods should exhibit an accelerated termination of photoresponse not only because of the shortened lifetime of active metarhodopsin II, but also due to a shortening of the activated PDE6 lifetime (Makino et al., 2004; Chen et al., 2012).
Modulation of the photoresponse by cGMP synthesis. A remarkable feature of the rod photoresponses is their amazing reproducibility (Gross et al., 2012a). Even shortening or extending the lifetimes of the activated metarhodopsin II or activated Tα-PDE6 complex do not exert substantial effects on the response amplitude (Chen et al., 2000; Krispel et al., 2003; Keresztes et al., 2004; Krispel et al., 2006). As discussed earlier, the amplitudes of the photoresponses in murine rods with an increased RGS activity, or rods deficient in Rgs9-1, Gβ5-L, or R9ap, are similar to wild-type rods. Also, extending the lifetime of active metarhodopsin II by knocking out Grk1 in mice exerts a rather small effect on the photoresponse amplitude; despite extending the lifetimes of active metarhodopsin II from ~40 ms in the wild-type rods to 76 ms, and even ~3 s in the knockout rods, the amplitudes of the photoresponses in the Grk1+/- and Grk1-/- rods increase only 1.2- and 1.7-fold, respectively, in comparison with the wild-type rods (Chen et al., 1999; Gross et al., 2012a). Consistent with this, shortening the lifetime of active metarhodopsin II to 15 ms in transgenic rods overexpressing GRK1 causes only a 24% decrease in the photoresponse amplitude (Gross et al., 2012a).
These results can be explained by considering that the kinetics of the photoresponse depend not only on the rate of cessation of metarhodopsin II activity, and the rate of cessation of PDE6 activity, which lead to cGMP hydrolysis, but also on the rate of synthesis of cGMP (Figure 4) (Gross et al., 2012a). Synthesis of cGMP is stimulated by a drop of [Ca2+], which occurrs upon the closure of cGMP-gated channels. While the influx of Ca2+ via cGMP-gated channels is inhibited, calcium ions are still being removed by the Na+/K+-Ca2+ exchanger, so the cytoplasmic [Ca2+] decreases (Figures 3, 4) (Cervetto et al., 1989; Schnetkamp, 2004). The decrease in [Ca2+] is sensed by proteins regulating the activities of the guanylate cyclases, which synthesize cGMP from GTP. Mammalian rods express two guanylate cyclases: GC1 (also known as GC-E) and GC2 (GC-F).
Guanylate cyclase activity is regulated by calcium-sensing proteins, which can either activate it when [Ca2+] decreases, or inhibit it when [Ca2+] is high. GC1 contains independent binding sites for four Ca2+-binding proteins: two activating proteins (guanylate cyclase activating protein 1 (GCAP1 and GCAP2), and two inhibitory proteins (S100B and neurocalcin) (Lange et al., 1999; Duda et al., 2002; Duda et al., 2005; Venkataraman et al., 2008). The mouse rod outer segment contains two of them: GCAP1 and GCAP2. In vitro, GCAP1 activates GC1 much more efficiently than GC2, whereas GCAP2 activates both GC1 and GC2 with similar efficiencies. The high free cytoplasmic [Ca2+], present in the dark, facilitates the binding of Ca2+ to GCAPs, thus preventing GCAPs from activating guanylate cyclases. Upon a decrease in the cytoplasmic [Ca2+], Ca2+ dissociates from GCAPs, so that GCAPs can activate guanylate cyclases to restore the cGMP level (Stephen et al., 2008). GCAP1 has a lower affinity for Ca2+ than does GCAP2, and therefore responds to a drop of cytoplasmic [Ca2+] more rapidly (Peshenko et al., 2011). The expression of only GCAP1 is enough to provide a response kinetics of rods to dim flashes of light similar to normal, but GCAP1-deficient rods saturate at lower light intensities (Howes et al., 2002; Makino et al., 2012). Further lowering of [Ca2+] leads to a dissociation of [Ca2+] from GCAP2, with a slower rate than from GCAP1. An expression of bovine GCAP2 in murine gcap−/− rods restores only partly the build-up of the response of rods to a dim flash of light, and the kinetics of recovery towards the photoresponse kinetics observed in the wild-type rods. Moreover, the GCAP2 expression in gcap−/− rods does not rescue the fast initial phase of recovery during light adaptation (Mendez et al., 2001).
Altogether, after an initial delay needed for the [Ca2+] decrease, and the dissociation of Ca2+ from GCAPs, the calcium-mediated feedback kicks in, leading to an increased GC activity, so that the rate of cGMP synthesis increases up to 10-fold. The stimulatory effect of GCAPs on guanylate cyclase activity occurs within ~40 ms after a short flash of dim light, that is, while the metarhodopsin II is still active. The calcium-mediated stimulation of cGMP synthesis narrows the variability of the photoresponses in wild-type rods, and can effectively oppose the effects of changing lifetimes of active metarhodopsin II or the Tα(GTP)-PDE6 complex, resulting from genetic manipulations (Gross et al., 2012a).
The importance of GCAPs in modulating photoresponses has been demonstrated by a comparison of the responses of the gcap-/- and wild-type rods to flashes of dim light (Burns et al., 2002). It has been shown that the amplitude of the circulating current in response to a dim flash is 5-fold smaller in the presence of GCAP than in its absence.
The Third Amplification Step: sensitivity of the cGMP-gated channel to cGMP. The cGMP-gated channels belong to the family of cyclic-nucleotide-gated channels, which are non-selective cation channels (Fu and Yau, 2007). In rods, the cGMP-gated channel is located on the plasma membrane of the outer segments, and it is a tetramer composed of three CNGA1 and one CNGB1 subunits. The CNGB1 subunit contains a binding site for the Ca2+-calmodulin complex (Grunwald et al., 1998, 1999; Weitz et al., 1998). The high cytoplasmic Ca2+ concentration in the dark results in the binding of Ca2+ to calmodulin, which then binds to the cGMP-gated channel, thereby decreasing its affinity for cGMP. This results in the greatest sensitivity of cGMP-gated channels to the cytoplasmic concentration of cGMP, which serves as the third step of amplification of phototransduction.
Altogether, the three amplification steps in phototransduction in dark-adapted rods enable the greatest sensitivity to light, so that visual perception occurs at irradiance levels, where only one or a couple of photons are absorbed in the rod outer segment. The responses of rods are characterized by an amazing reproducibility. However, the trade-offs for this high sensitivity of rods are: 1) the worsening of temporal resolution of vision because the phototransduction cascade is terminated slowly; and 2) reaching the saturation at a relatively low illuminance.
Light Adaptation in Rods
During an extended exposure to light, several changes occur in the rod leading to a partial recovery of the circulating current of cations. This is needed so that rods can avoid saturation, and can respond to light, albeit with decreased sensitivity. Importantly, in the light-adapted state, the temporal resolution is increased, so objects in motion can be followed more easily than in the dark-adapted state. Consideration of what happens at the molecular level during extended exposure to light allows some understanding of the mechanisms involved.
During constant exposure to light, the probability of photon absorption in the outer segment is decreased, because at least some rhodopsins are bleached. A fraction of bleached rhodopsins are present in the biochemically most active metarhodopsin II state, and each of them triggers a phototransduction cascade. As a result, an increased portion of cGMP channels becomes closed, thus disabling calcium ions to enter the photoreceptor. At the same time, the Na+/K+, Ca2+ exchangers remove cytoplasmic Ca2+ with a rate dependent on intra- and extracellular concentrations of Ca2+, K+ and Na+, and the polarization of the plasma membrane (Cervetto et al., 1989; Schnetkamp, 2004). When the free cytoplasmic concentration of Ca2+ decreases, it leads to the dissociation of calcium from Ca2+-binding proteins, such as recoverin, calmodulin, and GCAPs (Hsu and Molday, 1993; Hsu and Molday, 1994; Burns and Baylor, 2001; Mendez et al., 2001; Ames et al., 2006). Thus, the decrease of free cytoplasmic [Ca2+] provides a negative feedback activating processes responsible for the accelerated termination of phototransduction and the increased rate of cGMP synthesis, leading to a reopening of some cGMP-gated channels, which have been initially closed. As a result of the decreased free cytoplasmic [Ca2+], the amplification of phototransduction is decreased, which together with increased cGMP synthesis, worsens the sensitivity of rods.
The decrease in amplification observed upon an extended exposure to light is also due to a redistribution of recoverin, transducin, RGS9-1, Gβ5-L, and arrestin within the rod cell (Calvert et al., 2006; Tian et al., 2013). While in the dark-adapted state, rod outer segments contain the highest concentration of transducin (about 80-90% of total rod transducin) and recoverin (~12% of total rod recoverin), and the lowest concentration of RGS9-1, Gβ5-L and arrestin (less than 7% of total arrestin); during light adaptation these proteins translocate within minutes, but with different kinetics, into different parts of the rod cell. As a consequence, in the light-adapted state, only 10-20% and less than 2% of the total transducins and recoverins, respectively, are present in the outer segment (Calvert et al., 2006). In contrast, during an extended exposure to bright light, arrestin accumulates in the outer segment leading to an accumulation there of ~80% of total rod arrestin.
The reopening of a part of the cGMP-gated channels, which were initially closed, as well as the decreases in gains of the three amplification steps, are the main factors responsible for the adaptation of rods to light. They are discussed in more detail below.
The reopening of a part of the cGMP-gated channels, which were initially closed during exposure to light. As a result of the decrease in cytoplasmic concentration of cGMP, an increased fraction of cGMP-gated channels closes. The cytoplasmic concentration of cGMP is determined by the rate of cGMP hydrolysis by PDE6, and by the rate of synthesis of cGMP by guanylate cyclase (Dizhoor and Hurley, 1999; Haeseleer et al., 1999; Dizhoor et al., 2010; Gross et al., 2012a, 2012b). As mentioned earlier, an important effect, produced by the decrease in cytoplasmic [Ca2+], is an increased activity of guanylate cyclase, GC1 and GC2, caused by the dissociation of Ca2+ from GCAP1 and GCAP2. It has been estimated that a decrease in [Ca2+] in the light-adapted rods can increase guanylate cyclase activity 10-fold, in comparison to its activity in the dark (Koutalos et al., 1995; Burns et al., 2002; Makino et al., 2008). As a result of the increased concentration of cGMP, some closed cGMP-gated channels reopen, so that rods can recover some of the circulating current of cations, and therefore respond with a greater sensitivity to light stimuli.
Indeed, when a mouse rod is exposed to steady background light, the absolute value of the circulating current of cations decreases, but shortly after that, there is a partial recovery (Figure 6). The recovery follows biphasic kinetics; the fast component with a time constant of 380 ms, and a slow component with a time constant of ~84 s (Chen et al., 2010b). These changes in the circulating current correspond to the initial closure of cGMP-gated channels, followed by their partial reopening. The fast component of recovery is missing in a mouse rod deficient in GCAPs. This indicates that the fast component of the recovery is due to the time needed for the dissociation of Ca2+ from GCAPs, so they can activate GCs.
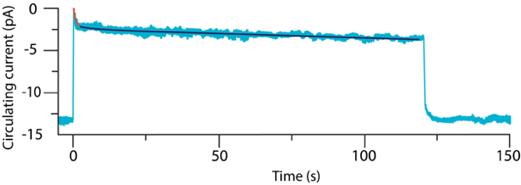
Figure 6. Change in the circulating current in the mouse rod exposed to light for 120 s. The initial, partial recovery of the circulating current is fitted to an exponential decay shown in red with a time constant of 0.38 s. The slow part of the recovery of the circulating current is fitted to an exponential decay shown in dark blue. (Modified from Chen et al., 2010b)
The effect of exposure to light on the first step in the amplification cascade. Exposure to light results in a decrease in free cytoplasmic [Ca2+], which subsequently affects the activities of several phototransduction-related proteins, including those responsible for setting the gain of the first step in the amplification cascade. Firstly, the decrease in free cytoplasmic [Ca2+] leads to a dissociation of Ca2+ from recoverin, which, in turn, causes a change in the recoverin conformation occurring on a millisecond timescale (Xu et al., 2011; Ames and Lim 2012). As a result, the myristoyl group becomes sequestered in a deep hydrophobic cavity inside the protein, where GRK1 used to be present when Ca2+ was still bound to recoverin. This change in conformation leads to a dissociation of recoverin from the lipid membrane and from GRK1. Upon release from the inhibitory effect of the Ca2+-recoverin complex, GRK1 can phosphorylate metarhodopsin II with an increased rate, thus competing with transducin for binding to metarhodopsin II, and eventually enabling binding of arrestin (Chen et al., 2010a). The arrestin binding stops completely the further activation of transducins by this metarhodopsin II. Therefore, the dissociation of Ca2+ from recoverin accelerates the inactivation of metarhodopsin II, so that the gain of the first amplification step of the phototransduction activated by that metarhodopsin II is decreased.
Moreover, studies in vitro suggest that the dissociation of Ca2+ from the Ca2+-calmodulin complex can lead to a dissociation of calmodulin from GRK1, thus relieving its inhibitory effect on the phosphorylation of metarhodopsin II (Grigoriev et al., 2012). Another potential effect of the dissociation of Ca2+ from calmodulin is the inactivation of AC1, followed by an inactivation of PKA, so it can no longer phosphorylate GRK1 (Osawa et al., 2011). It appears that the phosphorylation state of GRK1 is a result of a dynamic equilibrium between the kinase and phosphatase activities. When PKA activity decreases during exposure of the mouse retina to light, GRK1 is dephosphorylated with a time constant of 5.8 min. As a consequence, the kinase activity towards metarhodopsin II of the dephosphorylated GRK1 increases. All of these processes may also contribute to the decrease of the gain of the first amplification step in phototransduction.
The dissociation of recoverin from the disc membrane allows for its translocation from the outer segment into other parts of the rod. As a result of an extended exposure to bright light, the soluble recoverin moves towards the synaptic terminal, so that its concentration in the outer segment decreases about 6-fold (Strissel et al., 2005). The depletion of recoverin in the outer segment not only removes its inhibitory action on GRK1 (which does not undergo light-mediated redistribution), but may also decrease the Ca2+ buffering capacity of the rod outer segments (Chen et al., 2010a; Arshavsky and Burns, 2012). It has been suggested that recoverin can function as a powerful Ca2+ buffer (Reingruber et al., 2013). The concentration of free cytoplasmic Ca2+ in the dark-adapted mouse rod outer segments is estimated to be only ~0.3-0.6 µM, whereas the concentration of recoverin is ~600 µM, thus the 6-fold depletion of recoverin in the rod outer segments substantially reduces their Ca2+ buffering capacity. As a consequence, this may enable faster responses of other Ca2+-binding proteins, including calmodulin and GCAPs, when the closure of cGMP-gated channels leads to a decreased Ca2+ influx. On the other hand, exposure to bright light triggers the translocation of arrestin to the outer segment (Calvert et al., 2006). Arrestin can bind Ca2+ in a 1:1 stoichiometry, and therefore arrestin has also been suggested to act as a Ca2+ buffer (Huppertz et al., 1990).
The translocation of arrestin into the outer segment is another way to accelerate the inactivation of metarhodopsin II, and therefore to decrease the first amplification step of phototransduction. It has been estimated that only 1-7% of total rod arrestin is present in the dark-adapted outer segment, where it exists in a dynamic equilibrium of monomers, and self-associating dimers and tetramers (Schubert et al., 1999; Imamoto et al., 2003; Nair et al., 2005; Strissel et al., 2006; Hanson et al., 2007a, 2007b, Kim et al., 2011). Only the monomeric arrestin can bind to phosphorylated metarhodopsin II, whereas the oligomers serve as a storage, releasing arrestin monomers when needed to deactivate metarhodopsin II (Hanson et al., 2007a). It has been estimated that the concentration of free arrestin monomers is about 10 µM. The binding of monomeric arrestin to phosphorylated metarhodopsin II triggers the dissociation of oligomers, releasing more free monomers. Exposure to sufficiently bright light can eventually deplete the free arrestin in the outer segment, which then drives to the outer segment arrestin from other parts of rod cell (Strissel et al., 2006).
It has been suggested that the translocation of arrestin into the outer segment requires the microtubular cytoskeleton, and is initiated by phospholipase C (PLC), which activates PKC in the inner segment (Peterson et al., 2005; Reidel et al., 2008; Orisme et al., 2010). Pharmacological activation of PLC or PKC can trigger arrestin translocation even in the absence of light (Orisme et al., 2010). It is still unclear how PLC becomes activated in photoreceptors. Once activated, PLC hydrolyzes phosphatidylinositol-(4,5)-bisphosphate (PIP2) into diacyl glycerol (DAG) and inositol 1,4,5-triphosphate (IP3). The diffusable IP3 can bind to Ca2+ channels in the endoplasmic reticulum present in the inner segment, thereby triggering the release of Ca2+ to the cytoplasm. An increase in cytoplasmic Ca2+, together with DAG, can activate PKC. The activated PKC then phosphorylates the Bardet-Biedl syndrome 5 (BBS5) protein. BBS5 is a component of a supramolecular complex essential for the formation of primary cilia, including the photoreceptor outer segment (Smith et al., 2013). In the dark-adapted murine and frog photoreceptors, BBS5 is non-phosphorylated and is co-localized with arrestin along the microtubules of the axoneme in the outer segments, as well as in the inner segment and synaptic terminal. The phosphorylation of BBS5 reduces its affinity for arrestin, so that arrestin can dissociate and diffuse from the axoneme and inner segments towards the disc membranes, where it can interact with phosphorylated metarhodopsin II. It has been estimated that the movement of arrestin from the axoneme to the disc membranes happens on a millisecond timescale, whereas the translocation of arrestin from the inner segment to the outer segment takes up to 6-8 minutes, after which ~80% of total rod arrestin is accumulated in the outer segment (Elias et al., 2004).
The illuminance threshold above which the phosphorylation of BBS5 occurs, is the same for the massive translocation of arrestin into the outer segment. The threshold corresponds to the upper limit of the normal range of responsiveness of dark-adapted rods, which is reached when ~3% of rhodopsin is bleached (Strissel et al., 2006). Once the threshold is exceeded, the concentration of arrestin entering the outer segment can exceed 30 times the concentration of metarhodopsin II. The total rod arrestin accounts for about 0.8 mol eq of rhodopsin, indicating that there is enough arrestin to inactivate most metarhodopsin II/opsin even after an extensive bleach (Strissel et al., 2006; Hanson et al., 2007a). One arrestin can bind over a dimer composed of two phosphorylated metarhodopsins II, thus a single arrestin can prevent two molecules of metarhodopsin II from activating transducins (Sommer et al., 2012). Thus, even upon a complete bleach of all rhodopsins, there is more than enough arrestins to inactivate all phosphorylated metarhodopsins II/opsins.
The decrease of the gain of the first step of the amplification cascade, resulting from an extended exposure to bright light, is also due to the depletion of transducin in the outer segment. In the transducin molecule, the C-terminal of the γ subunit is farnesylated, and the N-terminal of the α subunit is acylated (Fu and Yau, 2007; Lobanova et al., 2010). These lipid modifications anchor the transducin heterotrimer to the disk membrane. Once Tα is separated from the transducin subunits β and γ (Tβγ), the bindings of these two individual parts to the lipid membrane are substantially weakened, so that each part can dissociate from the membrane. It has been suggested that the separated Tα is electrostatically repelled by the negatively charged membranes of rod disks (Lobanova et al., 2010).
Tα-GTP can be retained in the outer segment by binding with PDE6, which is strongly anchored to the disk membrane by isoprenylation of the C termini of both of its catalytic subunits (Catty et al., 1992; Qin and Baehr, 1994). However, PDE6 accounts for only ~0.1 mol eq of transducin, and therefore can easily get saturated upon exposure to bright light when a large proportion of the transducins becomes active. Indeed, it has been demonstrated that a significant translocation of transducin is observed only after exceeding a certain threshold of light intensity (Lobanova et al., 2007). The threshold also depends on the rate of deactivation of the Tα-PDE6 complex, which determines the concentration of free Tα and Tβγ. Once the threshold is exceeded, solubilised Tα and Tβγ diffuse, with slightly different kinetics, from the outer segment to the inner segment cell body and synaptic terminal (Sokolov et al., 2002; Kassai et al., 2005). Upon exposure to moderately bright light of 600 lx, the translocation of Tα is completed within 2 minutes, after which only 10-20% of Tα remains in the outer segment (Elias et al., 2004).
The decrease in transducin concentration in proximity of metarhodopsins II decreases the probability of transducin activation, and therefore decreases the amplification of phototransduction. It has been estimated that the translocation of transducin out of the outer segment can be responsible for almost a 10-fold reduction in the amplification of the signal (Sokolov et al., 2002).
The effect of exposure to light on the second step in the amplification cascade. Exposure to light seems to shorten the lifetime of the activated Tα(GTP)-PDE6 complex (Chen et al., 2012). As mentioned earlier, when a mouse rod is exposed to steady background light, the absolute value of the circulating current of cations decreases, but shortly after that, there is a partial recovery following biphasic kinetics, with the fast component corresponding to the activation of GCs with a time constant of 380 ms, and a slow component with a time constant of ~84s (Chen et al., 2010b). In the absence of GCAPs, the fast component is missing, whereas the time constant of the slow component is decreased to 28 s. It has been suggested that the slow component of the recovery is due to a decreased lifetime of the activated Tα(GTP)-PDE6 complex during an extended exposure to background light (Chen et al., 2010b; Korenbrot, 2012a; Korenbrot, 2012b). This suggestion is supported by the similarities between the photoresponses recorded from rods, preadapted to selected levels of background light, and then exposed to well defined flashes of light, and photoresponses obtained from mathematical modelling, based on an assumption that the Tα(GTP)-PDE6 lifetime decreases as a result of adaptation to the background light.
To explain the shortened lifetime of the activated Tα(GTP)-PDE6 complex, two potential mechanisms have been hypothesized. The first hypothesis stems from observations of the accelerated rates of termination of phototransduction in rods depleted from recoverin or overexpressing GRK1 (Chen et al., 2012; Grigoriev et al., 2012). As mentioned earlier, the accelerated rates can be explained assuming that GRK1 can accelerate the decay of the activated Tα(GTP)-PDE6 complex by the phosphorylation of one of its subunits Thus, all processes discussed in the preceding section that lead to activation of GRK1 during exposure to light: the dissociation of recoverin from GRK1 and its depletion in rod outer segment, the dissociation of calmodulin from RGK1, and the dephosphorylation of GRK1, may result in the GRK1-mediated phosphorylation of one or more proteins in the activated Tα(GTP)-PDE6 complex, such as the RGS9 components, transducin α, or PDE6 γ subunit.
In particular, the phosphorylation of the subunit γ of PDE6 has been considered as a possible mechanism responsible for that acceleration (Woodruff et al., 2008). The PDE6 subunit γ is a part of the GTPase activating complex, and therefore, it is tempting to speculate that its phosphorylation can accelerate the GTPase activity. This suggestion is supported by a comparison of photoresponses of wild-type rods with rods, in which the subunit γ of PDE6 lacks one of its phosphorylation sites due to a mutation causing threonine in position 35 to be replaced by alanine (T35A). Rods expressing PDE6 γ lacking this phosphorylation site exhibit almost no difference in the rate of photoresponse termination, independently of the background light (Woodruff et al., 2008). In contrast, in the wild-type rods, increasing the intensity of background light causes a shortening of the photoresponse termination after a flash of light.
However, rods with mutations in two phosphorylation sites, T22A and T35A in PDE6 γ subunit, exhibit light-adaptive changes in photoresponse termination similar to the wild-type rods (Woodruff et al., 2008). Moreover, upon an extended exposure to saturating light, the termination of phototransduction is accelerated, and exhibits similar rate constants for all three types of rods: wild-type rods and rods with single or double mutations, T22A and T35A. These results indicate that there are at least two different mechanisms involved in light-adaptive effects on the lifetime of active Tα(GTP)-PDE6. The role of GRK1 in the interaction with the GTPase complex needs further elucidation.
The second hypothetical mechanism responsible for the decreasing lifetime of the activated Tα(GTP)-PDE6 complex upon exposure to light, is the assembly of RGS9 in the outer segment (Tian et al., 2013). As mentioned earlier, in the dark-adapted rods, RGS9-1 and Gβ5-L are distributed outside of the outer segments, and RGS9-1 is heavily phosphorylated. It has been demonstrated that an exposure of the mouse retina to a light of illuminance of less than 1 lx is enough to initiate a translocation of both catalytic components of RGS9, RGS9-1 and Gβ5-L, towards the outer segment (Figure 4) (Tian et al., 2013). The translocation is very rapid; a 1 minute exposure to 20 lx is enough for a substantial redistribution of RGS9-1 towards the outer segment, whereas a 1 minute exposure to 150 lx leads to an accumulation of the majority of RGS9-1 in the outer segment. The translocation is thought to be triggered by the dephosphorylation of RGS9-1, which can be mediated by a decrease of cytoplasmic [Ca2+], which, in turn, can cause a decrease of PKC activity, and an increase of PP2A activity towards RGS9-1 (Balasubramanian et al., 2001; Hu et al., 2001; Sokal et al., 2003; Tian et al., 2013; Kirchhefer et al., 2014). The dephosphorylation of RGS9-1 increases its affinity to the R9AP present in the outer segment; so, together with Gβ5-L, they can assembly the RGS9 complex. RGS9 can then accelerate the GTPase activity of the activated Tα(GTP)-PDE6, thus decreasing the gain of the second amplification step in the phototransduction.
The RGS9 complex plays a major role in the termination of phototransduction and recovery of the circulating current after a flash of light. As mentioned earlier, in the absence of one of its components, the time constant of recovery of the circulating current upon an exposure of murine knockout rods to a dim flash of light is extended more than 10-fold in comparison with the wild-type rods (Chen et al., 2000; Krispel et al., 2003; Keresztes et al., 2004). Upon a saturating flash, the knockout rods remain in saturation considerably longer than the wild-type rods. The importance of the RGS9 components for human vision is underscored by the findings that mutations in RGS9-1 and R9AP affecting their functions cause their inability to adapt to changes in luminance, and to recognize moving objects (a condition known as bradyopsia) (Nishiguchi et al., 2004; Cheng et al., 2007; Hartong et al., 2007; Michaelides et al., 2010).
The effect of exposure to light on the third step in the amplification cascade. The dissociation of Ca2+ from calmodulin results in the dissociation of calmodulin from the cGMP-gated channel, which causes an increase in the affinity of the channels for cGMP, and therefore a decreased sensitivity of the cGMP-gated channels to changes in the concentration of cytoplasmic cGMP (Chen et al., 2010b). This may decrease the gain of the third step in the amplification cascade. It has been shown, however, that the photoresponse kinetics in mouse rods deficient in both GCAPs and the Ca2+-calmodulin binding site on the cGMP-gated channel, are similar to the photoresponse kinetics recorded for rods deficient only in GCAPs (Chen et al., 2010b). These observations suggest that the changes in affinity of the cGMP-gated channel for cGMP play only a minor role, in comparison with the effect of activation of guanylate cyclase by GCAPs.
The amplification factor in light-adapted rods. During constant exposure to background light, all factors discussed above: 1) the shortened lifetime of metarhodopsin II able to activate transducin, and smaller concentrations of transducins in the outer segment; 2) the shortened lifetime of active Tα(GTP)-PDE6; and 3) the increased affinity of cGMP-gated channels for cGMP, mean that fewer transducins can be activated by a single biochemically active metarhodopsin II, fewer cGMP molecules can be decomposed by a single active PDE, and the sensitivity of the channel to the cytoplasmic cGMP concentration is decreased (Burns and Pugh, 2010; Chen et al., 2010b). All of these factors contribute to the overall decrease in the amplification gain of the phototransduction cascade, and therefore to the loss of sensitivity to light. The diminished amplification is accompanied by a faster recovery of cGMP levels, which enable a better temporal resolution of vision, and extends the range of perceived illuminance levels. The trade-off here is an increased number of absorbed photons needed to elicit a detectable response in the light-adapted rods (Schneeweis and Schnapf, 2000).
Return of the Maximal Sensitivity of Rod after an Exposure to Light
Once the rod is exposed to light and then kept in the dark, several processes need to occur to restore the maximal sensitivity characteristic for its dark-adapted state: 1) all phototransduction cascades need to be terminated; 2) the concentration of cGMP, the ratio of open to closed cGMP-gated channels, and the circulating currents of Na+ and Ca2+ need to be restored; 3) the proteins that translocated during the exposure to light need to translocate back to restore their initial distribution in the dark; and 4) all phosphorylated metarhodopsins II with bound arrestins and other photobleached rhodopsins need to be dephosphorylated, dissociate from arrestins, and regenerate their dark-adapted configurations. These processes are interdependent and proceed through multiphasic kinetics.
An initial part of the recovery of the dark-adapted state occurs within hundreds of miliseconds up to several seconds, depending on how close to and how long in the saturation the photoresponse was. This fast part involves the inactivation of metarhodopsin II and the Tα(GTP)-PDE6 complex, as well as the activation of guanylate cyclases via mechanisms described in the preceding sections.
The inactivation of metarhodopsin II is facilitated by activated GRK1, which can rapidly phosphorylate metarhodopsin II as a result of preceding light-adaptation. Then, in the dark, the GRK1 activity is gradually diminished due to the influxes to the outer segment of Ca2+ and recoverin, as well as to GRK1 phosphorylation. In the mouse rods, the time constant of GRK1 phosphorylation in the dark, after a 2 hour exposure to light, is about 7 minutes (Osawa et al., 2011).
The complete inactivation of phosphorylated metarhodopsin II can be facilitated in the rod preadapted to bright light, because arrestin is accumulated in the outer segment. However, in the case of dark-adapted rods subjected to a rapid and massive bleaching of a substantial fraction of rhodopsin, which exceeds the concentration of arrestin available in the outer segment, the relocation of arrestin from the inner segment may become rate-limiting for the inactivation of metarhodopsin II, because it occurs on a timescale of several minutes (Elias et al., 2004). The insufficient deactivation by arrestin not only extends the lifetime of metarhodopsin II, but also allows the decay products of metarhodopsin II to activate transducin.
The inactivation of the Tα(GTP)-PDE6 complex is facilitated in the rod preadapted to light, due to the presence of the RGS complex in the outer segment. As a result of guanylate cyclase activation in the rod preadapted to light, the cytoplasmic concentration of cGMP increases, and subsequently more cGMP molecules bind to the cGMP-gated channels, so that an increased fraction of these channels open. This leads to influxes of Na+ and Ca2+ into the outer segment, gradually diminishing the polarisation of the plasma membrane. An increase in the free cytoplasmic [Ca2+] allows for the binding of Ca2+ to GCAPs so that they stop activating GC1 and GC2, and guanylate cyclase activities return towards their basal levels. The increased free cytoplasmic [Ca2+] also allows for the binding of Ca2+ to calmodulin, so that the calmodulin-Ca2+ complex can bind to the cGMP-gated channel, thereby increasing its sensitivity to cGMP (Rebrik et al., 2012).
Although the time constants of inactivation of metarhodopsin II and the Tα(GTP)-PDE6 complex in the dark-adapted mouse rods upon short flashes of light are about 40 ms and 200 ms, respectively, and both values decrease upon adaptation to light. The phototransduction cascades can persist in the dark on a time scale of several minutes or even tens of minutes. All these phototrasduction cascades need to be terminated before the cytoplasmic cGMP concentration is fully restored, the maximal fraction of the cGMP channels becomes open, the circulating current of cations is restored, and the polarisation of the rod plasma membrane decreases to the value characteristic for the dark-adapted state. These phototransduction cascades can be initiated in the dark by the products of rhodopsin bleaching, metarhodopsin III (possibly as a source of metarhodopsin II) and opsin, which can activate transducin, albeit with smaller efficiencies than metarhodopsin II (Okada et al., 1989; Fawzi and Northup, 1990; Cornwall and Fain, 1994; Melia et al., 1997; Nymark et al., 2012).
As mentioned earlier, the decay of metarhodopsins II and III occurs over a time course of several minutes, leading eventually to the hydrolysis of all-trans-retinal from the opsin active site (Ebrey, 1968; Cone and Cobbs, 1969; Baumann and Bender, 1973; Bennett, 1980; Nymark et al., 2012). The release of all-trans-retinal facilitates the dissociation of arrestin, and is followed by the dephosphorylation of opsin, at least in part by PP2A (Vishnivetskiy et al., 2007). All-trans-retinal remains non-covalently bound to opsin until it is reduced by retinol dehydrogenase 8 (RDH8) to all-trans-retinol, or until it is released from opsin when a newly delivered 11-cis-retinal regenerates rhodopsin (Heck et al., 2003b; Schadel et al., 2003) (see also: "Light-Induced Damage to the Retina").
These non-covalent complexes of all-trans-retinal with opsin can activate transducin, albeit with an efficiency of about 33 to 250 times smaller than the active metarhodopsin II (Jager et al., 1996; Surya and Knox 1997, 1998). To reduce the all-trans-retinal to all-trans-retinol, RDH8 requires NADPH as a hydrogen donor. NADPH is produced in the inner segment, and therefore needs to diffuse to the outer segment to be available for RDH8. The steady-state concentration of NADPH has been estimated to be about 1 mM in salamander rods (Miyagishima et al., 2009). Thus, an extensive bleaching of rhodopsin can lead to the depletion of NADPH in the outer segment, so that its delivery to the RDH8 in the outer segment limits the rate of all-trans-retinal reduction in amphibian rods (Tsina et al., 2004; Ala-Laurila et al., 2006; Kolesnikov et al., 2007; Miyagishima et al., 2009). Mammalian rods have a smaller outer segment volume than do salamander rods, and therefore the diffusion rate of NADPH may not be as limiting for the reduction of all-trans-retinal as it is for the salamander rods. Indeed, in contrast to salamander rods, where an extensive bleach is followed by the formation of all-trans-retinol initially only at the base of the outer segment, and then proceeds towards its distal end, a 1 minute exposure of isolated mouse rods to bleaching by light of >90% of rhodopsin is followed by a uniform formation of all-trans-retinol throughout the entire length of the outer segment with a time constant of about 17 min (Chen et al., 2009).
Opsin free of all-trans-retinal can also activate transducin, but its efficiency is about a million times smaller than that of metarhodopsin II (Cornwall and Fain, 1994; Surya et al., 1995; Melia et al., 1997). Considering that, upon an exposure to bright light, the bleaching products can be really abundant, they can provide a substantial contribution to the transducin activation and elevation of the rod photosensitivity threshold (Pepperberg et al., 1978; Pepperberg and Masland, 1978).
The activity of opsin bound with all-trans-retinal, or free opsin, can be quenched, similar to the case of metarhodopsin II, by phosphorylation and binding of arrestin. As mentioned previously, it is likely that PKC, rather than GRK1, is mainly responsible for the phosphorylation of opsin. The affinity of arrestin for the phosphorylated opsin is similar to its affinity for the phosphorylated metarhodopsin II.
The importance of deactivation of metarhodopsin II/opsin by the phosphorylation and binding of arrestin is underscored by an 8 to 10 times slower dark adaptation in persons with loss-of-function mutations in GRK1 or arrestin (Maeda et al., 2003; Lamb and Pugh, 2004). This condition, known as Oguchi disease, is also characterized by abnormalities in the recovery phase of the photoresponse kinetics, but the kinetics of rhodopsin regeneration are normal.
Importantly, the activity of opsin can be totally quenched by the binding of 11-cis-retinal, and regeneration of inactive rhodopsin. The availability of 11-cis-retinal for the regeneration of rhodopsin is dependent on the rate of its synthesis in the retinal pigment epithelium, and delivery to the rod outer segment. This multistep process in described by Rosalie Crouch in "The Visual Cycle", while certain aspects of it are discussed in more detail in "Light-Induced Damage to the Retina". Recent reviews provide our current understanding of the intricacies of the retinoid cycle and its role in the recovery of the dark-adapted state in rods (Lamb and Pugh, 2004, 2006; Kiser et al., 2012; Tang et al., 2013; Kiser et al., 2014).
The regeneration of rhodopsin in the human retina can be measured by reflection densitometry (Lamb and Pugh, 2004). It has been demonstrated that the regeneration of rhodopsin after a total bleach is completed within 15-20 min. However, a relatively short exposure to light of the human retina in vivo, such as 2 minutes, leads to a loss of photosensitivity of the rods, which then recovers in the dark following multiphasic kinetics over a longer timescale than expected based on the inactivation rate of metarhodopsin II/opsin (Figure 7) (Lamb and Pugh, 2004). For light exposures bleaching up to 10% of the rhodopsins, the fast component of rod recovery, with a time constant of 1.8 min, can be ascribed to the regeneration of bleached rhodopsins. For bleaches larger than 20%, the recovery is preceded by a delay proportional to the fraction of bleached rhodopsin up to a complete bleach of all rhodopsins. Following the initial delay, the fast component appears with the same time constant of 1.8 min. Then the recovery proceeds through a much slower time course with a time constant of about 6 min. Altogether, bleaching of >95% of rhodopsin is followed by about a 10 min delay before starting recovery, and in total, requires well over 30 minutes to recover the photosensitivity typical for the dark-adapted rods. It is still unclear what is responsible for setting the delay or for the slow component of recovery.
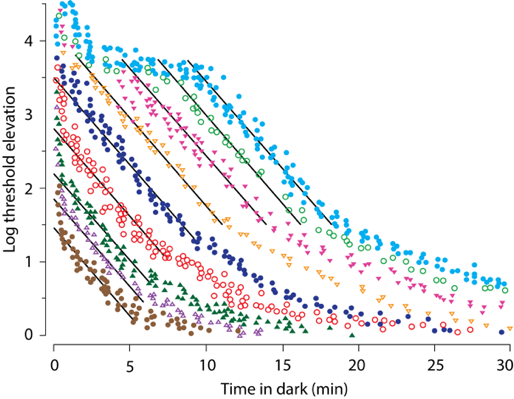
Figure 7. Recovery of photosensitivity in the dark, expressed as an elevation of the photosensitivity threshold, following flashes of light bleaching 0.5% (brown ● ), 2% (violet ∆), 4%(dark green ▲), 8% (red ○), 22% (dark blue ●), 39% (orange ∇), 63% (pink ▼), 86% (green ○), and 98% (cyan ●). The rapid phase of recovery reaches a plateau at ~3.7 log units, seen in bleaches of 39% and larger, due to the recovery of the cones. The black parallel lines are drawn close to the recovery component of rods with a time constant of 1.8 min. Note the delays in the recovery of rods for bleaches >20%, which increase with the increased percentage of bleached rhodopsin. Note also the slow component of the recovery, which is apparent for bleaches of 2% and more. Modified from Lamb and Pugh, 2004.
A slow process in the recovery of the dark-adapted state is the return of transducin to the outer segment (Elias et al., 2004; Zhang et al., 2011; Belcastro et al., 2012; Sinha et al., 2013). Thus, it suggests that the transducin translocation contributes to the slow component of the recovery of photosensitivity. After an extended exposure of mice to light, the return of transducin to the outer segment occurs in the dark with time constants of about 35 and 42 minutes for Tα and Tβγ, respectively. It appears, however, that these kinetics are strongly dependent on the fraction of bleached rhodopsin. Following a 10 min exposure to light that bleaches almost all of the rhodopsin, there is a substantial delay in the redistribution of transducin subunits in the dark (Belcastro et al., 2012). Thirty minutes of dark adaptation results in a very small increase of Tα in the outer segment, and no significant changes in the remaining parts of the rods. In contrast, a 10 min exposure to light that bleaches ~37% of the rhodopsin results in a substantial translocation of Tα towards the outer segment, clearly detectable after 30 minutes in the dark.
Several proteins present in rods have been proposed to facilitate the return of transducin to the outer segment; two of them are UNC119 and phosducin (Zhang et al., 2011; Belcastro et al., 2012; Sinha et al., 2013). UNC119 interacts with Tα, while phosducin has a high affinity for Tβγ.
UNC119 can interact with the myristoylated N-terminus of Tα(GTP) by internalization of the myristoyl chain into its binding pocket, which causes the dissociation of the Tα(GTP)-UNC119 complex from the lipid membrane (Zhang et al., 2011). Moreover, UNC119 inhibits the GTPase activity, and thereby provides a stable UNC119-Tα(GTP) complex, which can diffuse from the inner segment back to the outer segment. It is still unclear how quickly GTP in the complex is hydrolysed, and whether or not this is what triggers the dissociation of Tα(GDP) from UNC119.
The affinity of phosducin towards Tβγ can be regulated by phosphorylation (Belcastro et al., 2012; Sinha et al., 2013). In mouse rods, phosducin has two serines available for phosphorylation. The phosphorylation state of phosducin is increased in the dark, and regulated by the interplay of activity of PKA, Ca2+/calmodulin-dependent protein kinase (CaMKII) and PP2A. The phosphorylated phosducin has a decreased affinity for Tβγ, thus facilitates the translocation of Tβγ to the outer segment. The translocation is substantially delayed in mice expressing genetically modified phosducin without the phosphorylation sites, in comparison with mice expressing normal phosducin, which can undergo phosphorylation. Once in the outer segment, Tβγ recombines with Tα(GDP), thereby regenerating the inactive transducin trimer, which, once anchored to the disc membrane, stimulates the diffusion there of soluble subunits of transducin down the concentration gradient. Consistent with a need for an anchorage of the transduction trimer to drive the translocation, a deficiency of Tα in Gnat1-/- knockout mice, results in an inability of Tβγ to accumulate in the outer segment.
Conclusion
In conclusion, the understanding of the mechanisms responsible for adjusting the photosensitivity of rods to ambient light is growing. Increasingly sophisticated experiments have allowed the unravelling of intricate interactions between the proteins of rods, which, in turn, allow for substantial improvements in the mathematical modelling of processes responsible for the amazing adaptive changes in rods in response to light or lack thereof (Burns and Pugh, 2009; Gross et al., 2012b; Korenbrot, 2012b; Reingruber et al., 2013; Invergo et al., 2014).
Acknowledgements: Thanks to Ania Różanowska, Wojciech Różanowski and Marcin Zając for help in figure preparation.
References
Ala-Laurila, P, Kolesnikov, A V, Crouch, R K, Tsina, E, Shukolyukov, S A, Govardovskii, V I, Koutalos, Y, Wiggert, B, Estevez, M E and Cornwall, M C (2006). "Visual cycle: Dependence of retinol production and removal on photoproduct decay and cell morphology." J Gen Physiol 128(2): 153-169.
Ames, J B, Levay, K, Wingard, J N, Lusin, J D and Slepak, V Z (2006). "Structural basis for calcium-induced inhibition of rhodopsin kinase by recoverin." J Biol Chem 281(48): 37237-37245.
Ames, J B and Lim, S (2012). "Molecular structure and target recognition of neuronal calcium sensor proteins." Biochim Biophys Acta 1820(8): 1205-1213.
Arshavsky, V Y and Burns, M E (2012). "Photoreceptor signaling: Supporting vision across a wide range of light intensities." J Biol Chem 287(3): 1620-1626.
Baker, S A, Martemyanov, K A, Shavkunov, A S and Arshavsky, V Y (2006). "Kinetic mechanism of rgs9-1 potentiation by r9ap." Biochemistry 45(35): 10690-10697.
Balasubramanian, N, Levay, K, Keren-Raifman, T, Faurobert, E and Slepak, V Z (2001). "Phosphorylation of the regulator of g protein signaling rgs9-1 by protein kinase a is a potential mechanism of light- and Ca2+-mediated regulation of g protein function in photoreceptors." Biochemistry 40(42): 12619-12627.
Bartl, F J and Vogel, R (2007). "Structural and functional properties of metarhodopsin iii: Recent spectroscopic studies on deactivation pathways of rhodopsin." Phys Chem Chem Phys 9(14): 1648-1658.
Baumann, C and Bender, S (1973). "Kinetics of rhodopsin bleaching in the isolated human retina." J Physiol 235(3): 761-773.
Baylor, D A, Lamb, T D and Yau, K W (1979). "Responses of retinal rods to single photons." J Physiol 288: 613-634.
Belcastro, M, Song, H, Sinha, S, Song, C, Mathers, P H and Sokolov, M (2012). "Phosphorylation of phosducin accelerates rod recovery from transducin translocation." Invest Ophthalmol Vis Sci 53(6): 3084-3091.
Bennett, N (1980). "The decay of metarhodopsin ii in cattle rod outer segment membranes: Protonation and spectral changes." Biochem Biophys Res Commun 96(4): 1695-1701.
Burns, M E and Baylor, D A (2001). "Activation, deactivation, and adaptation in vertebrate photoreceptor cells." Annu Rev Neurosci 24: 779-805.
Burns, M E, Mendez, A, Chen, J and Baylor, D A (2002). "Dynamics of cyclic gmp synthesis in retinal rods." Neuron 36(1): 81-91.
Burns, M E and Pugh, E N, Jr. (2009). "Rgs9 concentration matters in rod phototransduction." Biophys J 97(6): 1538-1547.
Burns, M E and Pugh, E N, Jr. (2010). "Lessons from photoreceptors: Turning off g-protein signaling in living cells." Physiology (Bethesda) 25(2): 72-84.
Calvert, P D, Strissel, K J, Schiesser, W E, Pugh, E N, Jr. and Arshavsky, V Y (2006). "Light-driven translocation of signaling proteins in vertebrate photoreceptors." Trends Cell Biol 16(11): 560-568.
Catty, P, Pfister, C, Bruckert, F and Deterre, P (1992). "The cgmp phosphodiesterase-transducin complex of retinal rods. Membrane binding and subunits interactions." J Biol Chem 267(27): 19489-19493.
Cervetto, L, Lagnado, L, Perry, R J, Robinson, D W and McNaughton, P A (1989). "Extrusion of calcium from rod outer segments is driven by both sodium and potassium gradients." Nature 337(6209): 740-743.
Chen, C H, Blakeley, L R and Koutalos, Y (2009). "Formation of all-trans retinol after visual pigment bleaching in mouse photoreceptors." Investigative Ophthalmology & Visual Science 50(8): 3589-3595.
Chen, C K, Burns, M E, He, W, Wensel, T G, Baylor, D A and Simon, M I (2000). "Slowed recovery of rod photoresponse in mice lacking the gtpase accelerating protein rgs9-1." Nature 403(6769): 557-560.
Chen, C K, Burns, M E, Spencer, M, Niemi, G A, Chen, J, Hurley, J B, Baylor, D A and Simon, M I (1999). "Abnormal photoresponses and light-induced apoptosis in rods lacking rhodopsin kinase." Proc Natl Acad Sci U S A 96(7): 3718-3722.
Chen, C K, Woodruff, M L, Chen, F S, Chen, D and Fain, G L (2010a). "Background light produces a recoverin-dependent modulation of activated-rhodopsin lifetime in mouse rods." J Neurosci 30(4): 1213-1220.
Chen, C K, Woodruff, M L, Chen, F S, Chen, Y, Cilluffo, M C, Tranchina, D and Fain, G L (2012). "Modulation of mouse rod response decay by rhodopsin kinase and recoverin." J Neurosci 32(45): 15998-16006.
Chen, J, Makino, C L, Peachey, N S, Baylor, D A and Simon, M I (1995). "Mechanisms of rhodopsin inactivation in vivo as revealed by a cooh-terminal truncation mutant." Science 267(5196): 374-377.
Chen, J, Woodruff, M L, Wang, T, Concepcion, F A, Tranchina, D and Fain, G L (2010b). "Channel modulation and the mechanism of light adaptation in mouse rods." J Neurosci 30(48): 16232-16240.
Cheng, J Y, Luu, C D, Yong, V H, Mathur, R, Aung, T and Vithana, E N (2007). "Bradyopsia in an asian man." Arch Ophthalmol 125(8): 1138-1140.
Cone, R A and Cobbs, W H, 3rd (1969). "Rhodopsin cycle in the living eye of the rat." Nature 221(5183): 820-822.
Cornwall, M C and Fain, G L (1994). "Bleached pigment activates transduction in isolated rods of the salamander retina." J Physiol 480 ( Pt 2): 261-279.
Dizhoor, A M and Hurley, J B (1999). "Regulation of photoreceptor membrane guanylyl cyclases by guanylyl cyclase activator proteins." Methods 19(4): 521-531.
Dizhoor, A M, Olshevskaya, E V and Peshenko, I V (2010). "Mg2+/Ca2+ cation binding cycle of guanylyl cyclase activating proteins (gcaps): Role in regulation of photoreceptor guanylyl cyclase." Mol Cell Biochem 334(1-2): 117-124.
Doan, T, Mendez, A, Detwiler, P B, Chen, J and Rieke, F (2006). "Multiple phosphorylation sites confer reproducibility of the rod's single-photon responses." Science 313(5786): 530-533.
Duda, T, Fik-Rymarkiewicz, E, Venkataraman, V, Krishnan, R, Koch, K W and Sharma, R K (2005). "The calcium-sensor guanylate cyclase activating protein type 2 specific site in rod outer segment membrane guanylate cyclase type 1." Biochemistry 44(19): 7336-7345.
Duda, T, Koch, K W, Venkataraman, V, Lange, C, Beyermann, M and Sharma, R K (2002). "Ca(2+) sensor s100beta-modulated sites of membrane guanylate cyclase in the photoreceptor-bipolar synapse." EMBO J 21(11): 2547-2556.
Ebrey, T G (1968). "The thermal decay of the intermediates of rhodopsin in situ." Vision Res 8(8): 965-982.
Elias, R V, Sezate, S S, Cao, W and McGinnis, J F (2004). "Temporal kinetics of the light/dark translocation and compartmentation of arrestin and alpha-transducin in mouse photoreceptor cells." Mol Vis 10: 672-681.
Fain, G L (2011). "Adaptation of mammalian photoreceptors to background light: Putative role for direct modulation of phosphodiesterase." Mol Neurobiol 44(3): 374-382.
Fain, G L, Matthews, H R, Cornwall, M C and Koutalos, Y (2001). "Adaptation in vertebrate photoreceptors." Physiol Rev 81(1): 117-151.
Fawzi, A B and Northup, J K (1990). "Guanine nucleotide binding characteristics of transducin: Essential role of rhodopsin for rapid exchange of guanine nucleotides." Biochemistry 29(15): 3804-3812.
Fotiadis, D, Jastrzebska, B, Philippsen, A, Muller, D J, Palczewski, K and Engel, A (2006). "Structure of the rhodopsin dimer: A working model for g-protein-coupled receptors." Curr Opin Struct Biol 16(2): 252-259.
Fotiadis, D, Liang, Y, Filipek, S, Saperstein, D A, Engel, A and Palczewski, K (2003). "Atomic-force microscopy: Rhodopsin dimers in native disc membranes." Nature 421(6919): 127-128.
Fu, Y and Yau, K W (2007). "Phototransduction in mouse rods and cones." Pflugers Arch 454(5): 805-819.
Golobokova, E Y and Govardovskii, V I (2006). "Late stages of visual pigment photolysis in situ: Cones vs. Rods." Vision Res 46(14): 2287-2297.
Greene, N M, Williams, D S and Newton, A C (1995). "Kinetics and localization of the phosphorylation of rhodopsin by protein kinase c." J Biol Chem 270(12): 6710-6717.
Greene, N M, Williams, D S and Newton, A C (1997). "Identification of protein kinase c phosphorylation sites on bovine rhodopsin." J Biol Chem 272(16): 10341-10344.
Grigoriev, II, Senin, II, Tikhomirova, N K, Komolov, K E, Permyakov, S E, Zernii, E Y, Koch, K W and Philippov, P P (2012). "Synergetic effect of recoverin and calmodulin on regulation of rhodopsin kinase." Front Mol Neurosci 5: 28.
Gross, O P and Burns, M E (2010). "Control of rhodopsin's active lifetime by arrestin-1 expression in mammalian rods." J Neurosci 30(9): 3450-3457.
Gross, O P, Pugh, E N, Jr. and Burns, M E (2012a). "Calcium feedback to cgmp synthesis strongly attenuates single-photon responses driven by long rhodopsin lifetimes." Neuron 76(2): 370-382.
Gross, O P, Pugh, E N, Jr. and Burns, M E (2012b). "Spatiotemporal cgmp dynamics in living mouse rods." Biophys J 102(8): 1775-1784.
Grunwald, M E, Yu, W P, Yu, H H and Yau, K W (1998). "Identification of a domain on the beta-subunit of the rod cgmp-gated cation channel that mediates inhibition by calcium-calmodulin." J Biol Chem 273(15): 9148-9157.
Grunwald, M E, Zhong, H, Lai, J and Yau, K W (1999). "Molecular determinants of the modulation of cyclic nucleotide-activated channels by calmodulin." Proc Natl Acad Sci U S A 96(23): 13444-13449.
Gurevich, E V and Gurevich, V V (2006). "Arrestins: Ubiquitous regulators of cellular signaling pathways." Genome Biol 7(9): 236.
Haeseleer, F, Sokal, I, Li, N, Pettenati, M, Rao, N, Bronson, D, Wechter, R, Baehr, W and Palczewski, K (1999). "Molecular characterization of a third member of the guanylyl cyclase-activating protein subfamily." J Biol Chem 274(10): 6526-6535.
Hanson, S M, Gurevich, E V, Vishnivetskiy, S A, Ahmed, M R, Song, X and Gurevich, V V (2007a). "Each rhodopsin molecule binds its own arrestin." Proc Natl Acad Sci U S A 104(9): 3125-3128.
Hanson, S M, Van Eps, N, Francis, D J, Altenbach, C, Vishnivetskiy, S A, Arshavsky, V Y, Klug, C S, Hubbell, W L and Gurevich, V V (2007b). "Structure and function of the visual arrestin oligomer." EMBO J 26(6): 1726-1736.
Hartong, D T, Pott, J W and Kooijman, A C (2007). "Six patients with bradyopsia (slow vision): Clinical features and course of the disease." Ophthalmology 114(12): 2323-2331.
Heck, M, Schadel, S A, Maretzki, D, Bartl, F J, Ritter, E, Palczewski, K and Hofmann, K P (2003a). "Signaling states of rhodopsin. Formation of the storage form, metarhodopsin iii, from active metarhodopsin ii." J Biol Chem 278(5): 3162-3169.
Heck, M, Schadel, S A, Maretzki, D and Hofmann, K P (2003b). "Secondary binding sites of retinoids in opsin: Characterization and role in regeneration." Vision Res 43(28): 3003-3010.
Higgins, M K, Oprian, D D and Schertler, G F (2006). "Recoverin binds exclusively to an amphipathic peptide at the n terminus of rhodopsin kinase, inhibiting rhodopsin phosphorylation without affecting catalytic activity of the kinase." J Biol Chem 281(28): 19426-19432.
Howes, K A, Pennesi, M E, Sokal, I, Church-Kopish, J, Schmidt, B, Margolis, D, Frederick, J M, Rieke, F, Palczewski, K, Wu, S M, Detwiler, P B and Baehr, W (2002). "Gcap1 rescues rod photoreceptor response in gcap1/gcap2 knockout mice." EMBO J 21(7): 1545-1554.
Hsu, Y T and Molday, R S (1993). "Modulation of the cgmp-gated channel of rod photoreceptor cells by calmodulin." Nature 361(6407): 76-79.
Hsu, Y T and Molday, R S (1994). "Interaction of calmodulin with the cyclic gmp-gated channel of rod photoreceptor cells. Modulation of activity, affinity purification, and localization." J Biol Chem 269(47): 29765-29770.
Hu, G, Jang, G F, Cowan, C W, Wensel, T G and Palczewski, K (2001). "Phosphorylation of rgs9-1 by an endogenous protein kinase in rod outer segments." J Biol Chem 276(25): 22287-22295.
Huppertz, B, Weyand, I and Bauer, P J (1990). "Ca2+ binding capacity of cytoplasmic proteins from rod photoreceptors is mainly due to arrestin." J Biol Chem 265(16): 9470-9475.
Imai, H, Kefalov, V, Sakurai, K, Chisaka, O, Ueda, Y, Onishi, A, Morizumi, T, Fu, Y, Ichikawa, K, Nakatani, K, Honda, Y, Chen, J, Yau, K W and Shichida, Y (2007). "Molecular properties of rhodopsin and rod function." J Biol Chem 282(9): 6677-6684.
Imamoto, Y, Tamura, C, Kamikubo, H and Kataoka, M (2003). "Concentration-dependent tetramerization of bovine visual arrestin." Biophys J 85(2): 1186-1195.
Invergo, B M, Dell'orco, D, Montanucci, L, Koch, K W and Bertranpetit, J (2014). "A comprehensive model of the phototransduction cascade in mouse rod cells." Mol Biosyst.
Jager, S, Palczewski, K and Hofmann, K P (1996). "Opsin/all-trans-retinal complex activates transducin by different mechanisms than photolyzed rhodopsin." Biochemistry 35(9): 2901-2908.
Jastrzebska, B, Orban, T, Golczak, M, Engel, A and Palczewski, K (2013). "Asymmetry of the rhodopsin dimer in complex with transducin." FASEB J.
Kassai, H, Aiba, A, Nakao, K, Nakamura, K, Katsuki, M, Xiong, W H, Yau, K W, Imai, H, Shichida, Y, Satomi, Y, Takao, T, Okano, T and Fukada, Y (2005). "Farnesylation of retinal transducin underlies its translocation during light adaptation." Neuron 47(4): 529-539.
Keresztes, G, Martemyanov, K A, Krispel, C M, Mutai, H, Yoo, P J, Maison, S F, Burns, M E, Arshavsky, V Y and Heller, S (2004). "Absence of the rgs9.Gbeta5 gtpase-activating complex in photoreceptors of the r9ap knockout mouse." J Biol Chem 279(3): 1581-1584.
Kim, M, Hanson, S M, Vishnivetskiy, S A, Song, X, Cleghorn, W M, Hubbell, W L and Gurevich, V V (2011). "Robust self-association is a common feature of mammalian visual arrestin-1." Biochemistry 50(12): 2235-2242.
Kim, Y J, Hofmann, K P, Ernst, O P, Scheerer, P, Choe, H W and Sommer, M E (2013). "Crystal structure of preactivated arrestin p44." Nature 497(7447): 142-146.
Kirchhefer, U, Heinick, A, Konig, S, Kristensen, T, Muller, F U, Seidl, M D and Boknik, P (2014). "Protein phosphatase 2a is regulated by protein kinase calpha (pkcalpha)-dependent phosphorylation of its targeting subunit b56alpha at ser41." J Biol Chem 289(1): 163-176.
Kiser, P D, Golczak, M, Maeda, A and Palczewski, K (2012). "Key enzymes of the retinoid (visual) cycle in vertebrate retina." Biochim Biophys Acta 1821(1): 137-151.
Kiser, P D, Golczak, M and Palczewski, K (2014). "Chemistry of the retinoid (visual) cycle." Chem Rev 114(1): 194-232.
Knepp, A M, Periole, X, Marrink, S J, Sakmar, T P and Huber, T (2012). "Rhodopsin forms a dimer with cytoplasmic helix 8 contacts in native membranes." Biochemistry 51(9): 1819-1821.
Kohnken, R E, Chafouleas, J G, Eadie, D M, Means, A R and McConnell, D G (1981). "Calmodulin in bovine rod outer segments." J Biol Chem 256(23): 12517-12522.
Kolesnikov, A V, Ala-Laurila, P, Shukolyukov, S A, Crouch, R K, Wiggert, B, Estevez, M E, Govardovskii, V I and Cornwall, M C (2007). "Visual cycle and its metabolic support in gecko photoreceptors." Vision Res 47(3): 363-374.
Komolov, K E, Senin, II, Kovaleva, N A, Christoph, M P, Churumova, V A, Grigoriev, II, Akhtar, M, Philippov, P P and Koch, K W (2009). "Mechanism of rhodopsin kinase regulation by recoverin." J Neurochem 110(1): 72-79.
Korenbrot, J I (2012a). "Speed, adaptation, and stability of the response to light in cone photoreceptors: The functional role of ca-dependent modulation of ligand sensitivity in cgmp-gated ion channels." J Gen Physiol 139(1): 31-56.
Korenbrot, J I (2012b). "Speed, sensitivity, and stability of the light response in rod and cone photoreceptors: Facts and models." Prog Retin Eye Res 31(5): 442-466.
Kosloff, M, Travis, A M, Bosch, D E, Siderovski, D P and Arshavsky, V Y (2011). "Integrating energy calculations with functional assays to decipher the specificity of g protein-rgs protein interactions." Nat Struct Mol Biol 18(7): 846-853.
Koutalos, Y, Nakatani, K and Yau, K W (1995). "The cgmp-phosphodiesterase and its contribution to sensitivity regulation in retinal rods." J Gen Physiol 106(5): 891-921.
Kraft, T W, Schneeweis, D M and Schnapf, J L (1993). "Visual transduction in human rod photoreceptors." J Physiol 464: 747-765.
Krispel, C M, Chen, C K, Simon, M I and Burns, M E (2003). "Prolonged photoresponses and defective adaptation in rods of gbeta5-/- mice." J Neurosci 23(18): 6965-6971.
Krispel, C M, Chen, D, Melling, N, Chen, Y J, Martemyanov, K A, Quillinan, N, Arshavsky, V Y, Wensel, T G, Chen, C K and Burns, M E (2006). "Rgs expression rate-limits recovery of rod photoresponses." Neuron 51(4): 409-416.
Lamb, T D and Pugh, E N, Jr. (2004). "Dark adaptation and the retinoid cycle of vision." Prog Retin Eye Res 23(3): 307-380.
Lamb, T D and Pugh, E N, Jr. (2006). "Phototransduction, dark adaptation, and rhodopsin regeneration the proctor lecture." Invest Ophthalmol Vis Sci 47(12): 5137-5152.
Lange, C, Duda, T, Beyermann, M, Sharma, R K and Koch, K W (1999). "Regions in vertebrate photoreceptor guanylyl cyclase ros-gc1 involved in ca(2+)-dependent regulation by guanylyl cyclase-activating protein gcap-1." FEBS Lett 460(1): 27-31.
Leskov, I B, Klenchin, V A, Handy, J W, Whitlock, G G, Govardovskii, V I, Bownds, M D, Lamb, T D, Pugh, E N, Jr. and Arshavsky, V Y (2000). "The gain of rod phototransduction: Reconciliation of biochemical and electrophysiological measurements." Neuron 27(3): 525-537.
Levay, K, Satpaev, D K, Pronin, A N, Benovic, J L and Slepak, V Z (1998). "Localization of the sites for Ca2+-binding proteins on g protein-coupled receptor kinases." Biochemistry 37(39): 13650-13659.
Lobanova, E S, Finkelstein, S, Song, H, Tsang, S H, Chen, C K, Sokolov, M, Skiba, N P and Arshavsky, V Y (2007). "Transducin translocation in rods is triggered by saturation of the gtpase-activating complex." J Neurosci 27(5): 1151-1160.
Lobanova, E S, Herrmann, R, Finkelstein, S, Reidel, B, Skiba, N P, Deng, W T, Jo, R, Weiss, E R, Hauswirth, W W and Arshavsky, V Y (2010). "Mechanistic basis for the failure of cone transducin to translocate: Why cones are never blinded by light." J Neurosci 30(20): 6815-6824.
Maeda, T, Imanishi, Y and Palczewski, K (2003). "Rhodopsin phosphorylation: 30 years later." Prog Retin Eye Res 22(4): 417-434.
Makino, C L, Dodd, R L, Chen, J, Burns, M E, Roca, A, Simon, M I and Baylor, D A (2004). "Recoverin regulates light-dependent phosphodiesterase activity in retinal rods." J Gen Physiol 123(6): 729-741.
Makino, C L, Peshenko, I V, Wen, X H, Olshevskaya, E V, Barrett, R and Dizhoor, A M (2008). "A role for gcap2 in regulating the photoresponse. Guanylyl cyclase activation and rod electrophysiology in guca1b knock-out mice." J Biol Chem 283(43): 29135-29143.
Makino, C L, Wen, X H, Olshevskaya, E V, Peshenko, I V, Savchenko, A B and Dizhoor, A M (2012). " Enzymatic relay mechanism stimulates cyclic GMP synthesis in rod photoresponse: Biochemical and physiological study in guanylyl cyclase activating protein 1 knockout mice.” PLoS ONE 7(10): e47637.
Melia, T J, Jr., Cowan, C W, Angleson, J K and Wensel, T G (1997). "A comparison of the efficiency of g protein activation by ligand-free and light-activated forms of rhodopsin." Biophys J 73(6): 3182-3191.
Mendez, A, Burns, M E, Roca, A, Lem, J, Wu, L W, Simon, M I, Baylor, D A and Chen, J (2000a). "Rapid and reproducible deactivation of rhodopsin requires multiple phosphorylation sites." Neuron 28(1): 153-164.
Mendez, A, Burns, M E, Sokal, I, Dizhoor, A M, Baehr, W, Palczewski, K, Baylor, D A and Chen, J (2001). "Role of guanylate cyclase-activating proteins (gcaps) in setting the flash sensitivity of rod photoreceptors." Proc Natl Acad Sci U S A 98(17): 9948-9953.
Mendez, A, Krasnoperova, N V, Lem, J and Chen, J (2000b). "Functional study of rhodopsin phosphorylation in vivo." Methods Enzymol 316: 167-185.
Michaelides, M, Li, Z, Rana, N A, Richardson, E C, Hykin, P G, Moore, A T, Holder, G E and Webster, A R (2010). "Novel mutations and electrophysiologic findings in rgs9- and r9ap-associated retinal dysfunction (bradyopsia)." Ophthalmology 117(1): 120-127 e121.
Miyagishima, K J, Cornwall, M C and Sampath, A P (2009). "Metabolic constraints on the recovery of sensitivity after visual pigment bleaching in retinal rods." J Gen Physiol 134(3): 165-175.
Myers, W K, Xu, X, Li, C, Lagerstedt, J O, Budamagunta, M S, Voss, J C, Britt, R D and Ames, J B (2013). "Double electron-electron resonance probes ca(2)(+)-induced conformational changes and dimerization of recoverin." Biochemistry 52(34): 5800-5808.
Nair, K S, Hanson, S M, Mendez, A, Gurevich, E V, Kennedy, M J, Shestopalov, V I, Vishnivetskiy, S A, Chen, J, Hurley, J B, Gurevich, V V and Slepak, V Z (2005). "Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions." Neuron 46(4): 555-567.
Nikonov, S S, Brown, B M, Davis, J A, Zuniga, F I, Bragin, A, Pugh, E N, Jr. and Craft, C M (2008). "Mouse cones require an arrestin for normal inactivation of phototransduction." Neuron 59(3): 462-474.
Nishiguchi, K M, Sandberg, M A, Kooijman, A C, Martemyanov, K A, Pott, J W, Hagstrom, S A, Arshavsky, V Y, Berson, E L and Dryja, T P (2004). "Defects in rgs9 or its anchor protein r9ap in patients with slow photoreceptor deactivation." Nature 427(6969): 75-78.
Nymark, S, Frederiksen, R, Woodruff, M L, Cornwall, M C and Fain, G L (2012). "Bleaching of mouse rods: Microspectrophotometry and suction-electrode recording." J Physiol 590(Pt 10): 2353-2364.
Okada, D, Nakai, T and Ikai, A (1989). "Transducin activation by molecular species of rhodopsin other than metarhodopsin ii." Photochem Photobiol 49(2): 197-203.
Orisme, W, Li, J, Goldmann, T, Bolch, S, Wolfrum, U and Smith, W C (2010). "Light-dependent translocation of arrestin in rod photoreceptors is signaled through a phospholipase c cascade and requires atp." Cell Signal 22(3): 447-456.
Osawa, S, Jo, R, Xiong, Y, Reidel, B, Tserentsoodol, N, Arshavsky, V Y, Iuvone, P M and Weiss, E R (2011). "Phosphorylation of g protein-coupled receptor kinase 1 (grk1) is regulated by light but independent of phototransduction in rod photoreceptors." J Biol Chem 286(23): 20923-20929. Pepperberg, D R, Brown, P K, Lurie, M and Dowling, J E (1978). "Visual pigment and photoreceptor sensitivity in the isolated skate retina." J Gen Physiol 71(4): 369-396.
Pepperberg, D R and Masland, R H (1978). "Retinal-induced sensitization of light-adapted rabbit photoreceptors." Brain Res 151(1): 194-200.
Peshenko, I V, Olshevskaya, E V, Savchenko, A B, Karan, S, Palczewski, K, Baehr, W and Dizhoor, A M (2011). "Enzymatic properties and regulation of the native isozymes of retinal membrane guanylyl cyclase (retgc) from mouse photoreceptors." Biochemistry 50(25): 5590-5600.
Peterson, J J, Orisme, W, Fellows, J, McDowell, J H, Shelamer, C L, Dugger, D R and Smith, W C (2005). "A role for cytoskeletal elements in the light-driven translocation of proteins in rod photoreceptors." Invest Ophthalmol Vis Sci 46(11): 3988-3998.
Pugh, E N, Jr. (2006). "Rgs expression level precisely regulates the duration of rod photoresponses." Neuron 51(4): 391-393.
Qin, N and Baehr, W (1994). "Expression and mutagenesis of mouse rod photoreceptor cgmp phosphodiesterase." J Biol Chem 269(5): 3265-3271.
Rebrik, T I, Botchkina, I, Arshavsky, V Y, Craft, C M and Korenbrot, J I (2012). "Cng-modulin: A novel ca-dependent modulator of ligand sensitivity in cone photoreceptor cgmp-gated ion channels." J Neurosci 32(9): 3142-3153.
Reidel, B, Goldmann, T, Giessl, A and Wolfrum, U (2008). "The translocation of signaling molecules in dark adapting mammalian rod photoreceptor cells is dependent on the cytoskeleton." Cell Motil Cytoskeleton 65(10): 785-800.
Reingruber, J, Pahlberg, J, Woodruff, M L, Sampath, A P, Fain, G L and Holcman, D (2013). "Detection of single photons by toad and mouse rods." Proc Natl Acad Sci U S A 110(48): 19378-19383.
Rodieck, R W (1998). The first steps in seeing. Sunderland, MA, Sinauer Associates Inc.
Sampath, A P and Fain, G L (2009). "Setting the absolute threshold of vision." F1000 Biol Rep 1: 66.
Schadel, S A, Heck, M, Maretzki, D, Filipek, S, Teller, D C, Palczewski, K and Hofmann, K P (2003). "Ligand channeling within a g-protein-coupled receptor. The entry and exit of retinals in native opsin." J Biol Chem 278(27): 24896-24903.
Schneeweis, D M and Schnapf, J L (1995). "Photovoltage of rods and cones in the macaque retina." Science 268(5213): 1053-1056.
Schneeweis, D M and Schnapf, J L (1999). "The photovoltage of macaque cone photoreceptors: Adaptation, noise, and kinetics." J Neurosci 19(4): 1203-1216.
Schneeweis, D M and Schnapf, J L (2000). "Noise and light adaptation in rods of the macaque monkey." Vis Neurosci 17(5): 659-666.
Schnetkamp, P P (2004). "The SLC24 Na+/Ca2+-K+ exchanger family: Vision and beyond." Pflugers Arch 447(5): 683-688.
Schubert, C, Hirsch, J A, Gurevich, V V, Engelman, D M, Sigler, P B and Fleming, K G (1999). "Visual arrestin activity may be regulated by self-association." J Biol Chem 274(30): 21186-21190.
Siderovski, D P, Hessel, A, Chung, S, Mak, T W and Tyers, M (1996). "A new family of regulators of g-protein-coupled receptors?" Curr Biol 6(2): 211-212.
Sinha, S, Majumder, A, Belcastro, M, Sokolov, M and Artemyev, N O (2013). "Expression and subcellular distribution of unc119a, a protein partner of transducin alpha subunit in rod photoreceptors." Cell Signal 25(1): 341-348.
Smith, T S, Spitzbarth, B, Li, J, Dugger, D R, Stern-Schneider, G, Sehn, E, Bolch, S N, McDowell, J H, Tipton, J, Wolfrum, U and Smith, W C (2013). "Light-dependent phosphorylation of bardet-biedl syndrome 5 in photoreceptor cells modulates its interaction with arrestin1." Cell Mol Life Sci 70(23): 4603-4616.
Sokal, I, Hu, G, Liang, Y, Mao, M, Wensel, T G and Palczewski, K (2003). "Identification of protein kinase c isozymes responsible for the phosphorylation of photoreceptor-specific rgs9-1 at ser475." J Biol Chem 278(10): 8316-8325.
Sokolov, M, Lyubarsky, A L, Strissel, K J, Savchenko, A B, Govardovskii, V I, Pugh, E N, Jr. and Arshavsky, V Y (2002). "Massive light-driven translocation of transducin between the two major compartments of rod cells: A novel mechanism of light adaptation." Neuron 34(1): 95-106.
Sommer, M E and Farrens, D L (2006). "Arrestin can act as a regulator of rhodopsin photochemistry." Vision Res 46(27): 4532-4546.
Sommer, M E, Hofmann, K P and Heck, M (2012). "Distinct loops in arrestin differentially regulate ligand binding within the gpcr opsin." Nat Commun 3: 995.
Sommer, M E, Smith, W C and Farrens, D L (2006). "Dynamics of arrestin-rhodopsin interactions: Acidic phospholipids enable binding of arrestin to purified rhodopsin in detergent." J Biol Chem 281(14): 9407-9417.
Stephen, R, Filipek, S, Palczewski, K and Sousa, M C (2008). "Ca2+ -dependent regulation of phototransduction." Photochem Photobiol 84(4): 903-910.
Strissel, K J, Lishko, P V, Trieu, L H, Kennedy, M J, Hurley, J B and Arshavsky, V Y (2005). "Recoverin undergoes light-dependent intracellular translocation in rod photoreceptors." J Biol Chem 280(32): 29250-29255.
Strissel, K J, Sokolov, M, Trieu, L H and Arshavsky, V Y (2006). "Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin." J Neurosci 26(4): 1146-1153.
Surya, A, Foster, K W and Knox, B E (1995). "Transducin activation by the bovine opsin apoprotein." J Biol Chem 270(10): 5024-5031.
Surya, A and Knox, B E (1997). "Modulation of opsin apoprotein activity by retinal. Dark activity of rhodopsin formed at low temperature." J Biol Chem 272(35): 21745-21750.
Surya, A and Knox, B E (1998). "Enhancement of opsin activity by all-trans-retinal." Exp Eye Res 66(5): 599-603.
Tang, P H, Kono, M, Koutalos, Y, Ablonczy, Z and Crouch, R K (2013). "New insights into retinoid metabolism and cycling within the retina." Prog Retin Eye Res 32: 48-63.
Tian, M, Zallocchi, M, Wang, W, Chen, C K, Palczewski, K, Delimont, D, Cosgrove, D and Peng, Y W (2013). "Light-induced translocation of rgs9-1 and gbeta5l in mouse rod photoreceptors." PLoS One 8(3): e58832.
Tsina, E, Chen, C, Koutalos, Y, Ala-Laurila, P, Tsacopoulos, M, Wiggert, B, Crouch, R K and Cornwall, M C (2004). "Physiological and microfluorometric studies of reduction and clearance of retinal in bleached rod photoreceptors." J Gen Physiol 124(4): 429-443.
Venkataraman, V, Duda, T, Ravichandran, S and Sharma, R K (2008). "Neurocalcin delta modulation of ros-gc1, a new model of ca(2+) signaling." Biochemistry 47(25): 6590-6601.
Vishnivetskiy, S A, Raman, D, Wei, J, Kennedy, M J, Hurley, J B and Gurevich, V V (2007). "Regulation of arrestin binding by rhodopsin phosphorylation level." J Biol Chem 282(44): 32075-32083.
Wald, G and Brown, P K (1953). "The molar extinction of rhodopsin." J Gen Physiol 37(2): 189-200.
Wayman, G A, Impey, S, Wu, Z, Kindsvogel, W, Prichard, L and Storm, D R (1994). "Synergistic activation of the type i adenylyl cyclase by Ca2+ and gs-coupled receptors in vivo." J Biol Chem 269(41): 25400-25405.
Weitz, D, Zoche, M, Muller, F, Beyermann, M, Korschen, H G, Kaupp, U B and Koch, K W (1998). "Calmodulin controls the rod photoreceptor cng channel through an unconventional binding site in the n-terminus of the beta-subunit." EMBO J 17(8): 2273-2284.
Williams, D S, Liu, X, Schlamp, C L, Ondek, B, Jaken, S and Newton, A C (1997). "Characterization of protein kinase c in photoreceptor outer segments." J Neurochem 69(4): 1693-1702.
Woodruff, M L, Janisch, K M, Peshenko, I V, Dizhoor, A M, Tsang, S H and Fain, G L (2008). "Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors." J Neurosci 28(9): 2064-2074.
Xiong, W, Nakatani, K, Ye, B and Yau, K (1997). "Protein kinase c activity and light sensitivity of single amphibian rods." J Gen Physiol 110(4): 441-452.
Xu, X, Ishima, R and Ames, J B (2011). "Conformational dynamics of recoverin's Ca2+-myristoyl switch probed by 15n nmr relaxation dispersion and chemical shift analysis." Proteins 79(6): 1910-1922.
Zernii, E Y, Komolov, K E, Permyakov, S E, Kolpakova, T, Dell'orco, D, Poetzsch, A, Knyazeva, E L, Grigoriev, II, Permyakov, E A, Senin, II, Philippov, P P and Koch, K W (2011). "Involvement of the recoverin c-terminal segment in recognition of the target enzyme rhodopsin kinase." Biochem J 435(2): 441-450.
Zhang, H, Constantine, R, Vorobiev, S, Chen, Y, Seetharaman, J, Huang, Y J, Xiao, R, Montelione, G T, Gerstner, C D, Davis, M W, Inana, G, Whitby, F G, Jorgensen, E M, Hill, C P, Tong, L and Baehr, W (2011). "Unc119 is required for G protein trafficking in sensory neurons." Nat Neurosci 14(7): 874-880.
Zimmermann, K, Ritter, E, Bartl, F J, Hofmann, K P and Heck, M (2004). "Interaction with transducin depletes metarhodopsin III: A regulated retinal storage in visual signal transduction?" J Biol Chem 279(46): 48112-48119.
07/01/14