STRUCTURE and SPECTRA in BIOLUMINESCENCE
John Lee1 and Eugene S. Vysotski1,2
1Department of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602
jlee35uga@gmail.com
2Photobiology Lab, Institute of Biophysics, Russian Academy of Sciences (Siberian Branch), Krasnoyarsk 660036, Russia
eugene_vysotski@ibp.ru
In the accompanying modules on bioluminescence, we have seen how various organisms employ bioluminescence for their particular biological function. The bioluminescence color also has undergone adaptation and fine-tuning through the evolutionary process to enhance the host's survival potential. In this module, we will first consider the physical mechanisms by which a molecule becomes luminescent. In scientific terminology the first step is called "excitation" of the molecule, and this is followed by "emission" of a radiation characteristic of that molecule. In particular, we will describe mechanisms by which the color of the luminescence, called the "spectral distribution", can be modulated by the environment of the excited molecule. This then leads us to explanations of how protein structure plays its role in fine-tuning the color of bioluminescence emission.
THE FLUORESCENCE STATE
There are two main ways of obtaining the energy necessary to excite a molecule to luminesce, one is by absorbing the energy from light photons, and the other by the release of energy from a chemical reaction, known as "chemiluminescence". The common "light stick" is a familiar example of chemiluminescence. In order to get fluorescence, first of all the molecule has to be energized, and this is usually achieved through absorption of the energy contained in blue light or ultraviolet light. Absorption is a transformation of the light energy into excited electronic energy of the molecule. The fluorescence process is readily demonstrated by shining an ultraviolet lamp on some white clothing that has been washed in detergent. The ultraviolet energy is absorbed and re-emitted as blue (lower energy) fluorescence, because a blue fluorescing dye is commonly added to detergent so as to produce a wash that looks "whiter-than-white".
Bioluminescence is a chemiluminescence reaction in which a particular protein, called a luciferase or a photoprotein, is an essential requirement. The bioluminescence emission originates from the deposition of the reaction energy into the excited state of one of the participants in the biochemical system. This bioluminescence excited state is usually the same state responsible for fluorescence of that molecule and one of the aims in studying a bioluminescence mechanism is to identify the bioluminescence spectrum as belonging to the fluorescence of a reaction participant.
The energy contained in a photon of blue light (e.g., for wavelength in the 400 nm region) is given by the Planck equation:
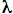
where h = Planck's constant, c = the velocity of light, and
= the wavelength of the light.
In order to consider photons as supplying energy to a chemical reaction we need to calculate the energy content of a "mole" of photons, called an "Einstein":
)
= (2.86 x 104 kcal-nm) / (400 nm)
= 71.5 kcal/mol.
This amount of energy, 71.5 kcal/mol, is a lot of energy to be obtained from a chemical process and is in the same range as the strength of covalent bonds. It certainly greatly exceeds the 7 kcal/mol of ATP hydrolysis free energy that biochemists normally regard as constituting a "high-energy" process. Oxidation reactions involving molecular oxygen however, are often capable of releasing this amount of energy, and it is therefore not surprising that all bioluminescence reactions do involve oxygen. But there is an obstacle that a chemical process has to get around if it is to be able to deposit this amount of energy into an excited state. Consider how the fluorescence state is achieved by the absorption of a photon. The photon field interacts with the electron cloud of the molecule with a certain probability that the result will be conversion of the photon into the molecule's excited electronic state, in other words absorption. This will occur in an exceedingly short time interval, a femtosecond
(10-15 s) or less. Excitation and emission steps are called "vertical" transitions, because only electrons can move in such short times, movements of atomic nuclei are frozen in this time interval (Visser and Rolinski, 2010). The problem in a theory of chemical excitation is that in the condensed state (liquid or solid), the chemical reaction energy release is produced by bond breakage and the shifting of atomic coordinates, occupying a time range up to a picosecond (10-12 s). In a picosecond time interval, molecular collisions would rapidly cool off excess energy and prevent it being accumulated, and available for vertical generation of excited states.
Several distinct processes have been considered to solve this difficulty, one is that energy release can be by some form of electron transfer. For example, if a covalent bond is cleaved homolytically to produce radicals, then the radical recombination and annihilation could be a vertical energetic process. The concerted decomposition of a highly energetic oxygen ring compound, called a dioxetanone, to yield radical intermediates, was first suggested (acronym "CIEEL") as the excitation mechanism for the firefly and other bioluminescences. An alternative picture currently favored, is consistent with the standard view of a chemical process where reactants are thermally activated to the transition state which has a structure close to that of the products. In chemical excitation this state would have the same electron distribution as the excited product and thus, adiabatic (no energy loss) crossing allows conversion to the co-ordinates of the excited state product molecule rather than its ground state. A high level computation based on the determined structure of firefly luciferase has been successfully applied to this case (Chung et al., 2008). Although computational approaches are currently an active area of research, a detailed discussion of this subject is beyond the scope of this module. In this section we will consider more qualitatively, the role of the protein structure in these processes.
ALTERATION OF ENERGY LEVELS
The Jablonski diagram (Figure 1) is a convenient way of illustrating the processes of excitation by absorption and the fluorescence emission from molecules. The radiative transitions are the straight vertical lines, the absorption event transforms the electronic state of the molecule from its ground state, S0, to a vibrationally excited "hot" state of the first excited singlet state S1', which cools rapidly (C, wavy line), a "non-adiabatic" change, to the semi-stable S1 level via collisional exchange with the environment.
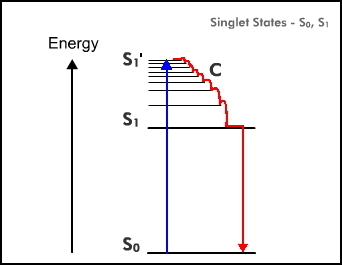
Figure 1. The Jablonski Diagram (simplified). Absorption (up arrow) is a rapid process (10-15 s), cooling (C, internal conversion) is slower (10-13 s) and fluorescence (down arrow) is much slower (10-9 s).
Whereas this absorption and cooling process is over in times much shorter than picoseconds as indicated, the S1 state is much more long-lived. Typical times before the fluorescence transition takes place are in the nanosecond (10-9 s) range. Also it must be remembered that in the condensed state, the transitions are not of a single energy but have a distribution of energies around an average; absorption and fluorescence spectra are broad bands usually about 100 nm width at half-height.
A consequence of excitation is a redistribution of electrons within the molecule so that the molecule in the S1 state almost always has a higher dipole moment than in its S0 state. Because the molecule is located in a "cage" of solvent molecules the energy level of S1 will include a contribution from electrostatic interaction of this dipole with the environment. From Coulomb's Law, this electrostatic energy will depend inversely on the dielectric constant so that in an aqueous solution this Coulombic contribution will be less than in a non-polar solvent. Consequently, the separation S1 - S0, will increase in environments of low dielectric constant (Figure 2). Experimentally it is found that the fluorescence spectrum of a compound in water is usually at a longer wavelength than when it is dissolved in an apolar solvent like hexane. The same argument can be extended to a fluorophore bound in a protein. If it is bound on the surface of the protein exposed to the aqueous surroundings, the fluorescence will be at a longer wavelength than if it were bound internally, in a hydrophobic cavity where the non-polar side chains of the amino acid residues predominate. Therefore, part of the effort made to identify the emitter in a bioluminescence system is to match the bioluminescence spectrum with the fluorescence of a candidate molecule in solvents of different dielectric constants.
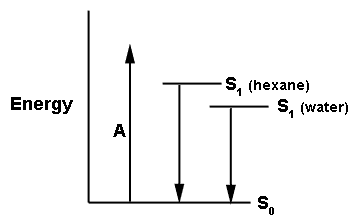
Figure 2. The effect of decrease in dielectric constant is to raise the energy of S1. The dielectric constant of water is 80 and hexane about 2. Consequently, the fluorescence spectrum of a molecule dissolved in hexane will be blue-shifted (higher energy) from that in water.
A second process that will perturb or change the energy level of the S1 state arises from the influence of the rigidity of the environment. In a rigid solvent such as glycerol at -20°C, fluorescence spectra shift to shorter wavelength. And so for a fluorophore rigidly bound within an enzyme active cavity, either itself tightly constrained via hydrogen bonding or alternatively, with the cavity restricting any adjustment of its conformation, then there might not be sufficient time for complete relaxation (R in Figure 3) before the fluorescence starts to occur and therefore the average fluorescence energy will increase, meaning that the fluorescence will be blue-shifted over that in a more fluid environment. Both of these conditions, binding site polarity or hydrophobicity, and rigidity, have to be considered in order to argue for the identification of a model fluorophore in solution as the emitting species in a bioluminescence reaction.
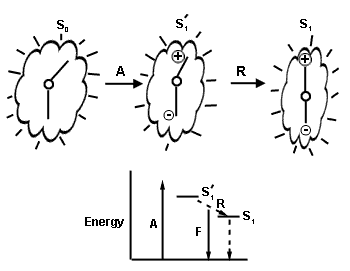
Figure 3. Rigidity also blue-shifts fluorescence spectra. The cartoon represents the fluorophore residing in the protein cavity where the surroundings restrict a change in the fluorophore's molecular structure (the two sticks). On absorption (A) to the initial excited state (S1') there is a redistribution of electrons resulting in an increase in dipole moment and tighter interactions with the surroundings. Relaxation (R) to the fluorescent state (S1) might be slowed enough that fluorescence (F) may occur before complete relaxation to S1. The S1 energy level itself will also be elevated by these interactions.
EMISSION FROM A SECOND MOLECULE
There are two further processes that can alter the fluorescence emission from a molecule in solution, and consequently can be used to explain the alteration of a bioluminescence spectrum due to environmental influences in a protein. Both these mechanisms have to occur within the nanosecond lifetime of the excited state of the initial product, and utilize a different fluorophore for the emission. The first is proton transfer, which can occur in times as short as the lifetime of the S1 state. If the initially excited molecule has acid-base properties, for example, it might possess a carboxyl or amine substitution, then proton transfer to or from a solvent molecule could quickly convert the initially excited state into an anion or cation excited molecule with changed spectral properties. Figure 4 is an example of the second possibility, "keto-enol tautomerism".
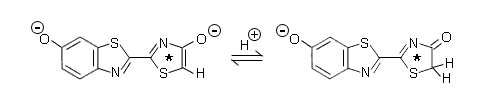
Figure 4. Keto-enol tautomerism in the excited state (*) of firefly oxyluciferin.
The initial product excited state of the firefly bioluminescence reaction is "oxyluciferin", the chemical structure on the right. A hydrogen attached to the carbon next to the carbonyl, migrates onto that carbonyl oxygen to change it to a hydroxyl group. Dissociation of this enol to the enolate on the left, produces the excited oxyluciferin anion with changed fluorescence properties. In the spatial structure of the active site of firefly luciferase in this case, to support this proposal one needs to identify a proximate base as the acceptor of the proton.
The second mechanism is one that makes use of a partner molecule and a process known as Förster Resonance Energy Transfer (FRET) (Visser and Rolinski, 2010). There are two well-studied examples of this in bioluminescence systems where the excitation generating protein, a luciferase or photoprotein, is associated with a second protein, descriptively named for its function as an "antenna protein". The antenna protein complexes with the luciferase in such a way that the electronic energy of the S1 state of the primary product undergoes resonance transfer to the antenna and the bioluminescence spectrum becomes identical to the fluorescence of the partner. The famous Green-Fluorescent Protein (GFP) is one such antenna protein identified as the bioluminescence emitter in many marine organisms. Another is "Lumazine Protein" (LumP) which is the origin of a deep blue color of many species of bioluminescent bacteria, and a closely related "Yellow-Fluorescent Protein" discovered in a yellow bioluminescence type of bacteria.
THE STRUCTURE OF BIOLUMINESCENCE PROTEINS
To obtain a high resolution spatial structure of a biological macromolecule is the "holy grail" of biochemistry research. Much expense is involved in building large machines such as high energy synchrotrons to provide an intense X-ray light source to attain this goal. Why are these structures so important to know? Consider how we might understand the mechanism of a chemical reaction. First we have to know the atomic structure of the reacting molecules, that is for example, that water is composed of two atoms of hydrogen and one of oxygen (known for more than 150 years), and that the O-H bonds subtend an angle of 105°, knowledge obtained by X-ray crystallography. This atomic structure explains why the water molecule has a net dipole moment. The structure then explains how the water molecule might react, say in hydrolyzing a molecule of ATP as we shall encounter in the firefly reaction, or participate in hydrogen bonding within or around a protein molecule.
The well-established methods of protein structure determination are by nuclear magnetic resonance and X-ray crystallography. The spatial structures of twenty or more types of bioluminescence proteins have been determined up to this time and can be viewed by accessing the Protein Data Bank (PDB). We will restrict ourselves here for brevity, to the examples of firefly luciferase, bacterial luciferase, calcium-regulated photoproteins, lumazine proteins, and varieties of GFPs. Several other structures available are for Renilla luciferase and Renilla coelenterazine binding protein, and dinoflagellate luciferase (Hastings et al., 2009).
An easy introduction to the use of crystallography for molecular structure determination is available on the internet: "X-ray Crystallography 101 Tutorial". Briefly, X-ray crystallography involves directing a narrow X-ray beam through a crystal, whereupon it is scattered by the electron clouds from each atom. A diffraction pattern on a detection plate results, and this is analyzed by highly developed computer programs utilizing "Fourier transformation", whereby the original electron density distribution of the target is fitted in an optimal way. The X-ray method is very powerful, but the main limitation is that a high quality single crystal of the macromolecule is first required, with dimensions on each side of at least 0.1 mm. In this regard many proteins have a tendency not to be cooperative. However, following the development of high expression systems with the ability to produce 10-100 mg of purified protein, extensive screening for crystallization conditions using efficient robotic devices has led to crystallization successes for an increasing number of proteins. Of particular concern to the subject here are the proteins listed above, and some others involved in these bioluminescence systems. We will discuss the photoproteins first, because of the particular depth of understanding of the bioluminescence reaction mechanism and spectra that has resulted from knowledge of their spatial structure.
THE CALCIUM-REGULATED PHOTOPROTEINS
These are called "calcium-regulated" photoproteins because the addition of Ca2+ triggers the bioluminescence emission, but is not involved in the chemical mechanism of light emission (Vysotski and Lee, 2004). No other additions are required to generate the bioluminescence, the reactive substrate bound with oxygen is already contained and stabilized within the purified photoprotein. In other words, the photoprotein can be regarded as a stabilized luciferase intermediate with Ca2+ just triggering its decomposition to produce a blue light emission. However, many of the organisms that contain photoproteins exhibit a green bioluminescence (in vivo) corresponding to the green fluorescence of GFP. This arises because the light organ (photophore) contains GFP weakly associated with the photoprotein, thus favoring FRET to the GFP.
Photoproteins are named for the marine organisms from which they are isolated, "aequorin" and "clytin" from the jellyfish, Aequorea and Clytia, and "obelin" from the hydroid Obelia. Many other bioluminescent marine organisms also employ photoproteins, but only these three have had their spatial structures determined. As these structures are highly similar, they are regarded as a sub-class of the also similar and far larger class populated by the many calcium-binding proteins.
The amino acid sequences of the photoproteins from six different species of Hydrozoa within the phylum Cnidaria, have been determined, and they are highly homologous, i.e., many of the amino acid residues occur in identical positions in each sequence, or the position is occupied by a similar residue, and this is usually taken to mean that these photoproteins arose from a common ancestor protein (Vysotski and Lee, 2004). Also on the basis of their primary sequence homology, they all would be expected to have the same three-dimensional structure.
The purified photoproteins have a yellow color due to the tightly bound reactive molecule, a 2-hydroperoxy derivative of "coelenterazine" (Figure 5).
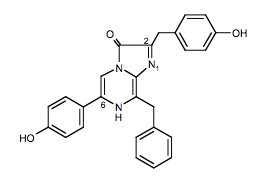
Figure 5. Coelenterazine: the active ingredient in the bioluminescence from coelenterates.
It is now realized that coelenterazine occurs in many marine organisms, even ones that are not bioluminescent, but its general function, other than for bioluminescence, is not known. Adding Ca2+ to a photoprotein produces a flash of light of duration tens to hundreds of milliseconds. It was unexpected to find in the first experiments carried out with aequorin, that oxygen was not required for the flash of bioluminescence, only Ca2+ (Shimomura, 2005). It did not matter whether the solution was air saturated or whether it was thoroughly degassed from oxygen, the flash was the same. At that time in the early 1960's, an oxygen involvement for all bioluminescence systems was believed to be the case and therefore it was proposed that oxygen must be already bound within the photoprotein along with the coelenterazine, and a suggestion that this substrate was a peroxide seemed reasonable (Figure 6). Following the determination of the chemical structure of coelenterazine, several lines of evidence, and most recently definitively from the three-dimensional structure (40 years later!), have confirmed this peroxide proposal. The overall reaction is now known to be an oxidative decarboxylation yielding carbon dioxide and excited coelenteramide as products (Figure 6).
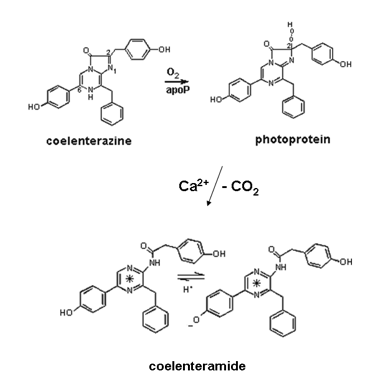
Figure 6. Overall chemical pathway for coelenterazine bioluminescence. In the upper reaction "apoP" means the "uncharged" protein. On binding coelenterazine, oxygen spontaneously adds to form the photoprotein-bound substrate, the 2-hydroperoxycoelenterazine (top right). Addition of Ca2+ triggers decarboxylation (-CO2) to produce the S1 state (*) of the bound product coelenteramide which can undergo excited state proton transfer to yield the S1 state of the phenolate (lower right structure).
Figure 7 shows the three-dimensional structure of the photoprotein obelin. All these photoproteins have a mass around 22 kDa and are mostly alpha helical in secondary structure composition, with four helix-turn-helix domains, labeled here I to IV. These structural domains are typical of the large family of calcium-binding proteins like calmodulin and troponin C. There are about 50 structures of such calcium-proteins available in the Protein Data Bank. In this protein family the site of calcium binding is always in the turn or loop regions. In the presence of Ca2+, the "discharged" photoproteins contain three bound calcium ions, each residing in the loops belonging to domains I, III and IV, but not II. The hydroperoxycoelenterazine substrate shown as the blue ball-and-stick model in Figure 7, is contained in a very hydrophobic pocket in the photoprotein, nestled between two of the helices.
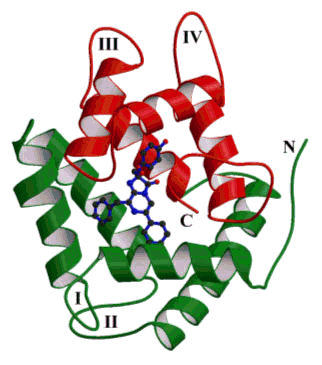
Figure 7. Structure of the Ca2+-regulated photoprotein obelin from Obelia geniculata (PDB code 1JF0). The N-terminal alpha-helices are green and the C-terminal red. The hydroperoxycoelenterazine substrate is the blue stick representation buried between green and red alpha-helices.
Figure 8 shows the electron density in the vicinity of the substrate binding pocket of obelin, revealed after analysis of its X-ray diffraction pattern. It is exceptional to obtain a protein structure at such high resolution. The chemical structure of the coelenterazine (Figure 7) fits accurately onto this density, except that there is extra density indicated by the blue arrow at the C-2 position of the coelenterazine ring, assignable as the two peroxy oxygens. This is the mentioned definitive evidence that the substrate exists in the protein as the peroxy derivative (Figure 6). Such a compound in free solution, however, is very unstable. In the protein in contrast, the oxygens are directed towards a tyrosine residue (Y190) with a separation between the last oxygen and the tyrosine's phenolic oxygen of 2.74 Å (1 Å = 0.1 nm). A distance in this range is indicative of a hydrogen bond leading to the conclusion that this strong hydrogen bond is the stabilizing factor. Photoproteins can therefore be regarded as a stabilized luciferase intermediate rather than as a separate class of bioluminescent protein.
Figure 9 is a flattened representation to help visualize how the amino acid residue side chains surround and interact with the hydroperoxycoelenterazine in the obelin cavity. Again the atom separations indicated by the dash lines and numbers (angstrom units; 1 Å = 0.1 nm), are interpreted to mean that the substrate is held in the site by hydrogen bonding to these residues. These dimensions shown here and the residue composition of the binding site, are almost the same for aequorin and obelin, yet their bioluminescence spectra differ.
The spectral maxima are 465 nm for aequorin, 475 nm for native obelin, and 485 nm and 495 nm for recombinant obelins from two species. The question of stabilization of the intermediate has been answered by making mutants at His179, Trp179, Tyr190, with single substitutions of residues with side groups having different H-bond properties (Eremeeva et al., 2013). All mutants lost their photoprotein property but most retained a luciferase-like activity, i.e., addition of coelenterazine to the apo-protein produced bioluminescence, a function not needing Ca2+. The chemistry of the light reaction is retained but the intermediate is not stabilized.
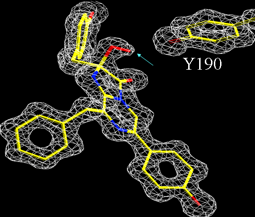
Figure 8. The electron density (white) in the binding pocket of obelin fits the known chemical structure (yellow) of coelenterazine except for extra density (blue arrow) at the 2-carbon, evidence for the peroxy (red) substituent, i.e., the 2-peroxy-coelenterazine. Nearby there is a tyrosine residue (Y190) from the protein at a distance indicating a hydrogen bond. This structure was determined at atomic resolution, 1.1 Å (see Liu et al., 2003).
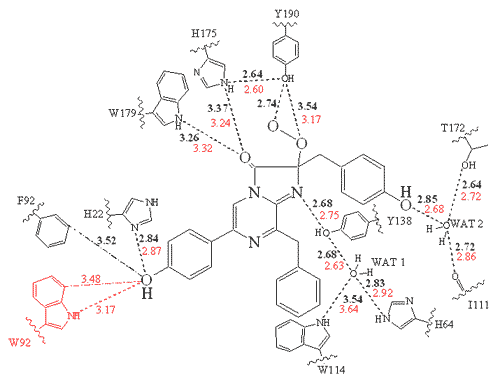
Figure 9. Two-dimensional representation of the hydroperoxycoelenterazine within obelin and its surrounding residues. The dotted lines indicate probable hydrogen bonds, and the numbers are the separations in Å units. The red numbers are for the obelin before the mutation of tryptophan 92 (W92) to phenylalanine (F92) (see Deng et al., 2001).
The question to answer then is: if the active site is the same among the photoproteins, how can the different bioluminescence spectra be explained? Also for the obelins, but not aequorin, the fluorescence maximum of the bound product, coelenteramide, is shifted about 25 nm to longer wavelengths than the bioluminescence maximum. The photoproteins are not fluorescent before reaction, fluorescence is developed only after the addition of Ca2+ to form the fluorescent product, the protein-bound coelenteramide. But we said that the bioluminescence excited state is the same as the fluorescence state, so how do we explain this anomaly?
We will make an "educated guess". An educated guess can be important in science if it leads to ideas about good experiments to test its validity. The thing we know is that the structures of the aequorin and obelin active sites are very similar but we cannot compare the two discharged photoproteins because only the Ca2+-discharged obelin structure has been solved (PDB code 2F8P). It is the environment of the excited coelenteramide that we need to compare in the two cases. We can get an idea about the effect of environment on free coelenteramide anion fluorescence by measurement under various solution conditions and it is found that the fluorescence spectral maximum can be shifted over a 460-520 nm range by change in solvent polarity. The educated guess then, based on model solution fluorescence studies, is that the effective dielectric constant of the coelenteramide binding site must be different among the Ca2+-discharged photoproteins, and different from the photoprotein itself. As the conformation of the binding site changes, the excited coelenteramide during its nanosecond fluorescence lifetime could experience a different environment depending on the time for relaxation of the cavity environment of that particular protein. The steady-state fluorescence spectrum reflects the influence of the final state environment, and the bioluminescence spectrum reflects that of various in-between states. The aim of experiments to validate these ideas will be to determine the spatial structure of Ca2+-discharged photoproteins, and to be able to trace the binding site dynamic changes.
Support for these ideas has already resulted from the fact that a tryptophan mutant of obelin changed the color of the bioluminescence from blue to violet. The tryptophan residue, W92 in Figure 9, is known from the obelin structure to be one forming part of the binding site, and it interacts via a hydrogen bond to the phenolic oxygen of coelenterazine. This is the one clear case for our bioluminescence proteins, where we can propose a structure-based explanation for the color shift. Figure 10 shows the bioluminescence spectrum from the obelin before and after the tryptophan mutation. The mutant produces to the eye, a violet bioluminescence, this color evidently due to the contribution of a second band appearing at shorter wavelength, the one with a maximum at 400 nm. In water, coelenteramide has no fluorescence, but becomes fluorescent in an aprotic solvent like dimethylsulfoxide, implying that the protein binding site environment might resemble such a solvent where water is excluded. Also, to make the fluorescence of free coelenteramide blue like the bioluminescence, the pH has to be made very basic, indicating that the blue fluorescing species is an anion of coelenteramide (Figure 6, lower right). If the solution is not basic the fluorescence is violet in color, just like the 400-nm bioluminescence band in Figure 10.
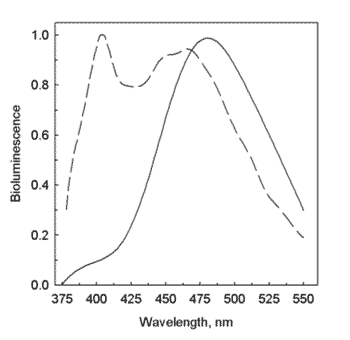
Figure 10. Bioluminescence spectrum from obelin (max. 485 nm; solid line) and after the tryptophan-92 to phenylalanine mutation (max. 400, 460 nm; dashed line) (see Deng et al., 2001; Vysotski et al., 2003).
How does this shorter wavelength band develop in the bioluminescence protein after the Trp92 is substituted with phenylalanine? Notice in Figure 9 that, nearby the oxygen of coelenterazine there is a histidine (H22) as well as the Trp92, both in a hydrogen bond interaction with that phenolic oxygen. These interactions remain unchanged in the structure of the Ca2+-discharged obelin. A significant outcome from the study that determined this last structure was a postulate that the decarboxylation should result in the primary product being the neutral excited state of coelenteramide (Figure 6, lower left). Donation of the H-bond from the Trp92 deposits positive charge onto the acceptor oxygen, consequently enhancing rapid transfer of the phenolic proton to His22, leaving the phenolate still in its excited state. It was proposed that the blue bioluminescence color therefore originates from this excited phenolate in an ion-pair interaction with H22 (Figure 9). Removing the H-bond to Trp92 slows the rate of proton transfer allowing time for the primary excited product, the neutral coelenteramide to radiate from its S1 state. As this has a higher energy a violet bioluminescence is seen. That the proximate H22 controls this process in some way is indicated by substitution of H22 by glutamate, resulting in a completely monomodal band of violet bioluminescence (Figure 11; violet, 370 nm maximum). Green emission from Y138F shows that hydrogen bonding at N1 also controls the color in some way. Again, we need to determine spatial structures of these mutants if we want to have more insight into how these various hydrogen bonds control these processes.
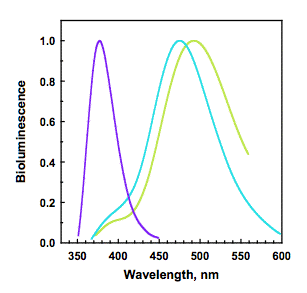
Figure 11. Bioluminescence of obelin: wild-type (cyan); and mutants W92F-H22E (violet) and Y138F (green) (see Frank et al., 2008).
In Figure 12 it is seen that in aequorin there is a hydrogen bond to the bound coelenterazine's phenolic oxygen from a tyrosine residue (Tyr82), which is absent in obelin (right), as the equivalent sequence position is a phenylalanine (Phe88). The difference in spectra between aequorin and obelin is qualitatively attributable to the Tyr82 hydrogen bond, because both bioluminescence and fluorescence are interchanged by switching these residues. Figure 13A shows for obelin (dashes), substitution of its Phe88 by Tyrosine to provide a third hydrogen bond shifts the bioluminescence to shorter wavelength (maximum 453 nm). In contrast, for aequorin (Figure 13B, dashes) the reverse happens, Tyr82 for phenylalanine shifts the bioluminescence to longer wavelength, maximum 501 nm. Hydrogen bonding at this position is solely in control of the spectra because the spatial structures of the obelin mutants F88Y before and after bioluminescence, are closely homologous to the corresponding obelin structures (Natashin et al., 2014). Figure 14 illustrates that the bioluminescence color from photoproteins may be tuned from the violet to green by the application of such "rational" mutations.
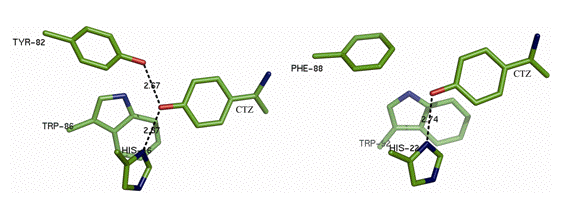
Figure 12. Hydrogen bonds (dashed line) to the phenolic oxygen (red) of coelenterazine (CTZ) in aequorin (left) and obelin (right) (see van Oort et al., 2009).
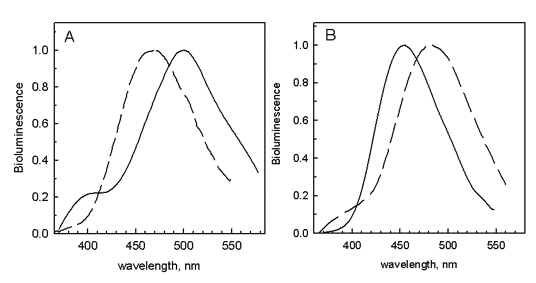
Figure 13. The bioluminescence spectra of aequorin (A) and obelin (B) are interchanged by a switch of hydrogen bonding in the active site by substituting a Phe for Tyr, and vice versa, respectively (see Stepanyuk et al., 2005).
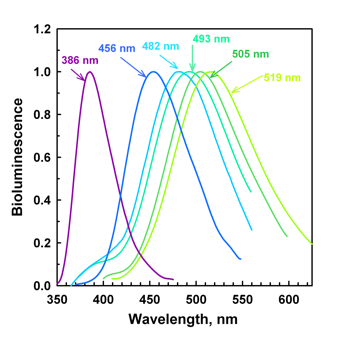
Figure 14. Obelin bioluminescence "rainbow" resulting from rational mutations of binding site residues (see Malikova et al., 2003; Stepanyuk et al., 2005; Frank et al., 2008) . The spectral maxima are labeled.
FIREFLY LUCIFERASE
Firefly bioluminescence was the first system to receive extensive investigation with modern biochemical techniques (Lee, 2014). By the end of the 1950s, it had been established that the in vitro bioluminescence of firefly required ATP, Mg ion, molecular oxygen, an enzyme called firefly luciferase, and a small organic molecule substrate, firefly luciferin. The luciferase and luciferin were obtained in highly purified form, and the chemical structure of the firefly luciferin (LH2) was solved (Figure 15).
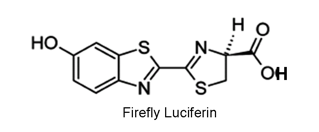
Figure 15. Firefly D-luciferin (LH2) is used by all bioluminescent beetles. Only the D-isomer (indicated by the dash and triangular bonds) is active.
The bioluminescence quantum yield, defined as the number of photons (or einsteins) emitted divided by the number of luciferin molecules (or moles) consumed, for firefly luciferin was in 1959, the first bioluminescence yield to be measured, and was found to have an astonishingly high value of 0.88 (± 0.28). This number has recently been more accurately redetermined to be in the range 0.4-0.6 (± ~0.1), depending on the type of firefly luciferase, but it still remains the highest quantum yield found for a bioluminescence system. The chemistry of the light reaction was completely established by the early 1970's. It occurs in several steps indicated by the double arrows below, the first being the formation of a luciferyl-AMP intermediate. Oxygen addition to this is followed by oxidative decarboxylation to products CO2 and the excited state (*) of oxy-Luciferin (Branchini, 2013; Vieira et al., 2012). Note the overall mechanistic similarity to the photoprotein chemistry (Figure 6), also involving a decarboxylation. In the overall equation below, AMP is adenosine monophosphate and PPi is inorganic pyrophosphate.
LH2 + ATP + O2 → → CO2 + oxy-Luciferin* + AMP + PPi
The in vitro reaction carried out at the optimal pH 7.8, produces a yellow bioluminescence, a broad spectrum with maximum at 562 nm, nearly the same as the in vivo bioluminescence. The fluorescence of the reaction products is also yellow, similar to the bioluminescence, and this led to the early conclusion that the oxy-Luciferin was the bioluminescence light emitter. However, a puzzling finding was that the bioluminescence spectrum could be shifted from yellow to red under the influence of various agents, such as the solution pH lowered to around 6, increase of temperature over 40°C, or addition of Cd2+ or Hg2+. It was proposed that the excited oxy-Luciferin product could exist either as the keto-phenolate (Figure 4, right) or after keto-enol tautomerism, the enol-phenolate, one structure having a lower energy S1 level (red) than the other (yellow). Positive identification of the bioluminescence emitters from fluorescence measurements was frustrated at that time, by the instability of oxy-Luciferin, and the difficulty in its purification.
These first studies of the firefly mechanism used mainly extracts from the North American firefly, Photinus pyralis. The firefly is not a true fly (Diptera) but a beetle, belonging to the order Coleoptera. Other bioluminescent insects within this order are the Caribbean click beetle and the South American railroad-worm. All beetle luciferases use the same substrates, firefly D-luciferin, ATP (Mg2+), and oxygen. However, their bioluminescence spectra depend on the type of organism, and range from the familiar yellow color of many fireflies, to red for the railroad-worm, maximum 623 nm (Viviani, 2009). The luciferases isolated from the different beetles produce in vitro bioluminescence spectra corresponding to the in vivo color of that beetle. Also, the different luciferases all have strong sequence similarity, leading to an expectation that the overall spatial structures of all the beetle luciferases should be similar. Color mutants can also be produced and, in one example, the substitution of a single residue shifts the bioluminescence maximum from 550 nm to 620 nm. This means that the color variations must arise from differences in interactions of the excited oxy-Luciferin with residues comprising the binding site of the beetle luciferase (Viviani et al., 2013). In the last few years, structural investigations of both players, the oxy-Luciferin and the firefly luciferase, now allow atomic level rationalization for the origin of this bioluminescence color variation.
The critical breakthrough has been in the development of new purification techniques for oxy-Luciferin (Naumov et al., 2009). As a result the crystal structure of this key molecular participant has been determined and its spectral properties precisely characterized. In the crystal, oxy-Luciferin exists in the trans configuration and forms head-to-tail dimers of the phenol-enol form (Figure 16, top-left). Presumably this configuration is the one of lowest energy, probably due to extensive hydrogen bonding, which can be expected to apply also to oxy-Luciferin residing in the luciferase binding site. Figure 15 shows the six molecular forms of oxy-Luciferin that would be expected to be in equilibrium over the physiological pH range 6-8.5. The neutral forms have a blue fluorescence, which eliminates them as candidates for the bioluminescence emitter. The anions have fluorescence spectra spanning the green through red range of bioluminescence colors, and these spectra are shifted depending on solvent properties, pH, polarity, H-bonding, etc.
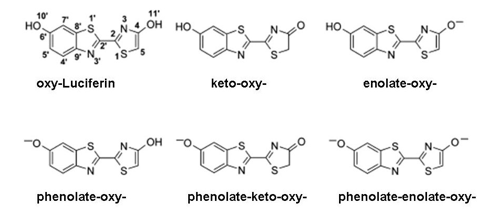
Figure 16. Six ionic forms of oxy-Luciferin. Crystal structure shows that the trans structure of the keto-oxy-Luciferin is the most stable form (see Naumov et al., 2009).
The red bioluminescence is tentatively attributed to the phenolate-keto-oxy-Luciferin from the observation that oxy-Luciferin shows a corresponding red fluorescence spectrum under basic conditions, and that a red bioluminescence is found using a 5,5'-dimethyl-luciferin derivative, where the product, 5,5'-dimethyl-oxy-Luciferin cannot undergo keto-enol tautomerism (Branchini et al., 2002) (Figure 17). In the crystal, this 5,5'-dimethyl oxy-Luciferin is verified to exist in the keto-oxyluciferin form, and would be expected to dissociate to the phenolate-keto-oxyluciferin form at physiological pH, arguing for this anion as the origin of the red bioluminescence. The green to orange range of bioluminescence cannot be assigned to a particular oxy-Luciferin anion as they all should exist in equilibrium in aqueous solution at pH 6-8.5. In fact, over this range the fluorescence spectra are found to be composed of mixtures. It is concluded that any of the anionic forms, including the phenolate-keto form, could be favored by various environmental interactions giving rise to the green to orange range of beetle bioluminescence color.
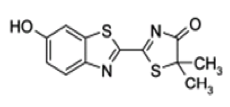
Figure 17. 5,5'-Dimethyl-oxy-Luciferin, which cannot undergo keto-enol tautomerism, is the red bioluminescence emitter in the reaction with 5,5'-dimethyl firefly luciferin as substrate.
Figure 18 shows the 3D-structure of firefly luciferase from Luciola cruciata, a species common to Japan. Firefly luciferase is much larger than the photoproteins just described, being a 62-kDa single chain protein with a distinctly different topology. It is comprised of a large domain at the N-terminal (Figure 18, gray) and a smaller one (Figure 17, blue) at the C-terminal, with beta sheets interspersed with short alpha helices. Firefly luciferase is structurally related to an enzyme of quite unrelated metabolic function, a peptide synthetase that catalyzes the ATP activation of a precursor in the synthetic pathway to a cyclic polypeptide, Gramicidin S. They both have the ATP activation step in common, going to a carboxyl-AMP product, but in the case of firefly luciferase, this product, luciferyl-AMP, spontaneously adds molecular oxygen.
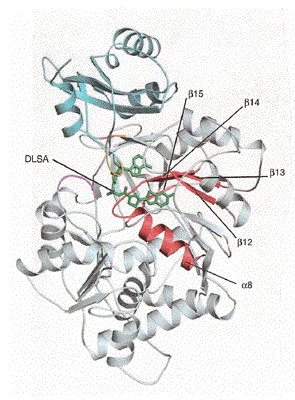
Figure 18. Three-dimensional structure of firefly luciferase (PDB code 2D1S). An analog of the luciferin-AMP, DLSA, is located in the active site (green stick structure) (see Nakatsu et al., 2006).
This luciferase structure has been obtained containing an analog of firefly luciferin, DLSA, so that we can say that the location of the firefly luciferin and ATP substrate binding sites in the structure of firefly luciferase, is now known with reasonable certainty. Obviously the luciferin itself would not be useable for this purpose, so the protein crystal studied was grown with this DLSA, an unreactive analog of the luciferyl-AMP residing in the binding site (Figure 19). The active site in any enzyme structure in most cases, has to be located in a like manner, by including with the crystal a substrate analog or a competitive inhibitor, because the substrate itself of course, could not persist there, because the property of the active site is to catalyze the reaction rapidly to product. We have seen, however, that photoproteins are a fortunate exception, as they have bound the stabilized reaction intermediate, the peroxidized substrate. An alternative approach for locating the active site, which was also applied in the first studies of firefly luciferase, was to draw on the structural homology to a peptide synthetase that carries out an analogous chemistry producing phenylalanine adenylate. From these various lines of evidence we can be reasonably certain of the location of the active site in firefly luciferase. A two-dimensional picture of the luciferin-AMP analog in the binding site is in Figure 19.
A second firefly luciferase structure determined was with the genuine reaction products oxy-Luciferin and AMP, and they were found to reside in the same binding cavity as the luciferin (PDB code 2D1R; Figure 20). The configuration of these products was practically the same as for the luciferin-AMP analog, but there were movements indicated by the arrows, of some residues into the cavity and overall, the various shifts create a more hydrophobic environment for the oxy-Luciferin. Thus, changes in hydrophobicity and/or ionic equilibria can rationalize the green to orange spread of the firefly bioluminescence colors. There are many residue substitutions which affect the bioluminescence color, some quite far from the active cavity. Substitutions in the loop residues 220-235, which are nearby but not in contact with the substrate, have strong effects. It was suggested that these might open up the cavity to expose the excited product to an interaction with solvent. Direct evidence for this possibility is in a third structure (PDB code 2D1T) obtained for mutated luciferase S285N, which produces red bioluminescence with firefly luciferin. The vicinity of the oxy-Luciferin is more open than in the unsubstituted luciferase, more exposed to solvent and with a more polar surrounding, conditions favoring a red shift of fluorescence spectra.
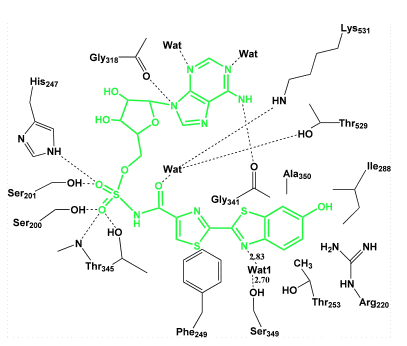
Figure 19. Two-dimensional diagram of the binding sites in firefly luciferase. A luciferin-AMP analog (green structure) placed in the center is seen to form hydrogen bonds (dots) with some binding site residues (black), and to fixed water (Wat) molecules (see Nakatsu et al., 2006).
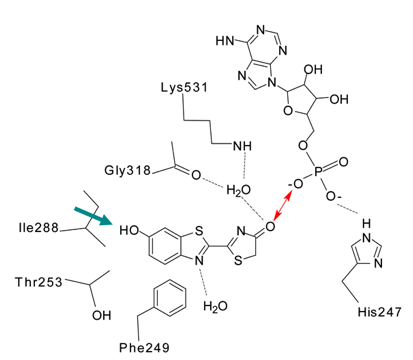
Figure 20. Two-dimensional diagram of firefly luciferase with oxy-Luciferin and AMP in the binding cavity. The red arrow suggests an ionic interaction at the keto position. The side chain of Isoleucine288 has moved closer to the phenol end (green arrow) than before reaction (Figure 17) (see Naumov et al., 2009).
BACTERIAL LUCIFERASE
This is the third class of bioluminescence system where the biochemistry has been well studied. However, in spite of many research efforts, the reaction mechanism of bacterial bioluminescence has not yet been elucidated. Unlike the firefly and coelenterazine chemistry, there is no CO2 product, and it is generally believed that the bacterial mechanism must be completely different (Lee, 2014). The bacterial bioluminescence substrates are reduced flavin mononucleotide (FMNH2), a fatty aldehyde like tetradecanal, and oxygen. Bacterial luciferase is completely unrelated to firefly luciferase, the photoproteins, or other coelenterazine luciferases, both in terms of primary sequence and spatial structure. Bacterial luciferase has a mass of 77 kDa, and consists of two similar, but non-identical, subunits, called alpha and beta. The three-dimensional structure (Figure 21) shows a lot of alpha helices and the flavin binding site resides in a groove formed within beta barrels of the alpha subunit. There are many types of bioluminescent bacteria, and they utilize the same substrates and have homologous luciferase primary sequences, implying that they will have the same spatial structures. Although they have no relationship to other luciferases other than the property of light emission, the bacterial luciferases together with a number of flavo-enzymes quite unrelated in terms of their biochemistry, have been found to have homologous spatial structures, leading to their grouping into a "bacterial luciferase family" of TIM-barrel proteins. This lends additional support to the proposal of "convergent evolution", that all bioluminescent systems have arrived at their light emitting function through different evolutionary pathways, not from a common ancestor.
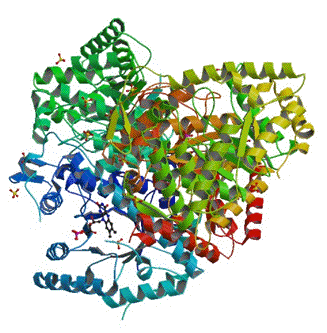
Figure 21. Structure of bacterial luciferase (PDB code 3FGC). It consists of two similar, but not identical subunits, alpha (blue and dark green) and beta (red, brown, and light green). The small ring structure at the 7 o'clock position, is the FMN located in a cavity on the alpha subunit (see Campbell et al., 2009).
Bacterial luciferases isolated from the different types of bacteria, show a smaller range of bioluminescence spectral maxima than photoprotein or beetle bioluminescence, i.e., only from 490 nm to 509 nm (Lee et al., 1990). One final product of the reaction is the fully oxidized substance FMN whose fluorescence maximum is at 525 nm in water. There are no solution conditions which shift this maximum very much to shorter wavelength towards a match for the bioluminescence; FMN fluorescence is shifted to slightly longer wavelengths in solvents of low dielectric constant. During the bioluminescence reaction however, an intermediate is observed having the same fluorescence spectrum as the bioluminescence emission. This intermediate is proposed to be a protein-bound 4a-hydroxy-FMNH, which subsequently would dehydrate to FMN after release from the luciferase. Unfortunately it is difficult to study the 4a-hydroxyflavin itself due to its instability in solution, but analogs have been prepared and they have weak fluorescence in this region with spectral sensitivity to solvent polarity, so variations in effective dielectric properties of the binding site could explain the small bioluminescence spectral variation among the different bacterial luciferases. Unfortunately, a structural comparison of binding sites among luciferases cannot be done, because only one type of bacterial luciferase structure has been determined, that from the species Vibrio harveyi.
THE ANTENNA PROTEINS
A. Lumazine protein in bacterial bioluminescence. In some bioluminescence systems spectral shifts are achieved by means of an accompanying fluorescent protein, named for its function as an "antenna protein". The bioluminescent bacteria for example, exhibit a range of in vivo bioluminescence spectral maxima 470-542 nm, much larger than for the in vitro reaction with different bacterial luciferases. The shortest in vivo wavelength maximum, 470 nm appears to be characteristic for species belonging to the genus Photobacterium. The longest spectral maximum is 545 nm, a yellow bioluminescence found in a single strain of Alliivibrio sifiae (previously called Vibrio fischeri, strain Y1) (Figure 22).

Figure 22. Liquid flask cultures of glowing bioluminescent bacteria. Left: Photobacterium species. Right: Y1 strain of Alliivibrio sifiae (see Daubner et al., 1987).
The reason for the wider range of bioluminescence from the bacteria themselves was shown to be due to the presence of a novel type of fluorescent antenna protein that associates with the luciferase. Bacterial luciferase and the antenna protein form a protein-protein complex in which the excited state of the primary excited reaction product is coupled to the electronic transition of the proximate fluorophore bound within the antenna protein (Titushin et al., 2011). It was found that the antenna proteins purified from either the blue or the yellow types were very similar. Both have a mass of 21 kDa, and homologous amino acid sequences, with the difference being only in the nature of the fluorophore. The antenna protein from Photobacterium is called "Lumazine Protein", because it has as a fluorescent ligand, 6,7-dimethyl-8-ribityl-lumazine (Figure 23) (Lee et al., 1990). In equations lumazine protein is given the symbol "LumP". This lumazine derivative is highly fluorescent, and when bound to the protein its spectrum is identical to the 470 nm bioluminescence. The homologous yellow-fluorescent antenna protein from the yellow bacterium has a flavin as the ligand. The ligands are non-covalently bound and can be switched in either protein, flavin for the lumazine, and vice versa.
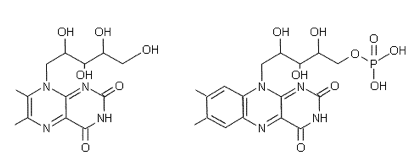
6,7-dimethyl-8-ribityl-lumazine (left) and
Flavin Mononucleotide (FMN) (right).
The fluorescence properties of ligands bound in lumazine proteins may be rationalized by examination of spatial structure (Figure 24). The lumazine derivative is buried in a hydrophobic pocket which would result in the blue shift of its fluorescence maximum from 490 nm in water to 470 nm bound in the protein cavity. If the lumazine is replaced by FMN, the structure displays the ribityl-phosphate tail sticking out into solution, providing an enhanced dipole moment and rationalizing the unusual flavin fluorescence shift to longer wavelength, from its green maximum at 525 nm in water to yellow at 545 nm in the protein.
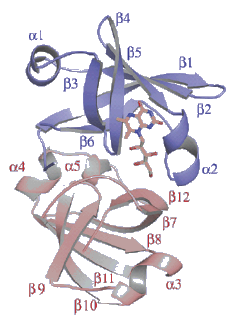
Figure 24. Structure of lumazine protein from Photobacterium. It consists of two similar, but non-identical domains, indicated by the blue and pink helices. The lumazine ligand is the yellow stick model (see Sato et al., 2010).
Another curiosity is that the yellow protein is strongly fluorescent in contrast to the weak fluorescence, if any, of flavoproteins in general. Even more remarkable is that the lumazine derivative happens to be a precursor in the biosynthetic pathway of flavins, specifically it is the substrate of riboflavin synthetase, which is present in many bacteria, and there is in fact, both sequence and structural homology between lumazine protein and the enzyme riboflavin synthetase. It has been suggested that there has been a "borrowing" of proteins from one function to suit another purpose, in the process of the evolution of bacterial bioluminescence (O'Kane et al., 1991).
There is a considerable amount of biophysical evidence to indicate that the mechanism causing these bioluminescence color shifts in the bacteria is by Förster Resonance Energy Transfer (FRET), via a weak dipole-dipole electronic coupling between the luciferase-produced excited state, the "fluorescent transient" fluorophore, and the ligand of the antenna protein (Petushkov et al., 1996). Dynamic fluorescence experiments showed that there was a complex formed between bacterial luciferase and lumazine protein, and with the recent availability of their spatial structures, a computationally docked model of this complex has been produced (Figure 25). A separation of only 10 Å between the two fluorophores, provides a very favorable circumstance for the proposed Förster mechanism of bioluminescence color shift.
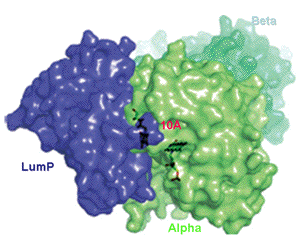
Figures 25. Lumazine protein (LumP, blue) associated to the alpha subunit (green) of bacterial luciferase (Alpha, green: Beta, pale green) revealing a 10 Å separation between the lumazine binding site on LumP and the flavin-active site of luciferase (see Sato et al., 2010).
B. Green-fluorescent protein in some marine bioluminescence systems. The Green-fluorescent protein (GFP) is probably the most famous protein in Science. This is not because of GFP's function in bioluminescence, where it was first identified, but because of the myriad applications of GFP as a fluorescent marker in studies of gene expression, cell imaging, and clinical diagnostics. These aspects are the subject of another module (Zimmer, 2010). Here we will describe how GFP was recognized as a participant in bioluminescence, and what is known about its mechanism of function as an antenna protein (Ward, 1998).
In 1962, the active photoprotein aequorin that had been purified from extracts of the jellyfish Aequorea, was observed to produce a blue (λmax 465 nm) bioluminescence unlike the green (λmax 510 nm) emission from the animal. In that report it was noticed that there was a green protein component in extracts of the jellyfish, and by the early 70's, evidence was accumulating for the presence of GFP's in a number of different marine organisms, such as the hydroid Obelia and the sea pansy Renilla (Figure 26). It was proposed that the antenna function of GFP operates by the Förster resonance energy transfer mechanism but the details still remain obscure to this day. In the case of renilla GFP the formation of a complex with renilla luciferase was reported in the late 70's (Ward, 1998). The spatial structures of many types of GFPs have been determined, both ones from bioluminescent organisms as well as the related ones that have some function other than bioluminescence, such responsible for the coloration of corals (Matz et al., 2002).
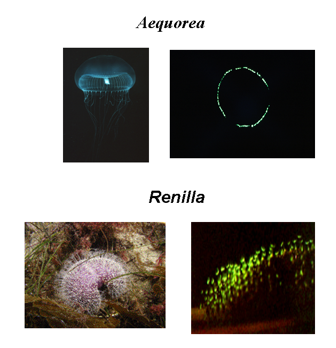
Figure 26. The presence of Green-Fluorescent Proteins in several marine organisms is the origin of the green color of the in vivo bioluminescence. The photos show the jellyfish Aequorea and the sea-pansy Renilla in room light (left), and the bioluminescence in the dark (right).
The GFP's occurring in bioluminescent organisms all have fluorescence maxima in the range only 497-509 nm. The range for the coral GFPs extends into the red. In the reaction of a photoprotein or renilla luciferase, inclusion of GFP in concentrations of only micromolar, already reveals a shift of the bioluminescence spectrum towards the fluorescence of GFP itself (Figure 27). The effect is specific for the type of GFP, i.e., renilla bioluminescence is shifted by renilla GFP, but not by aequorea GFP. For the FRET mechanism to operate, there must be a protein-protein complex formed, but such a stable complex has not been observed at micromolar concentrations. Instead, a very weak complex is detected at millimolar concentrations, so it is postulated that the FRET must occur in a transiently formed complex in the course of the reaction. The possible structure of this complex has been obtained by computational docking of the known spatial structures of the component proteins (Figure 28). It would have a tetrameric configuration, and as such, would allow a very efficient FRET to occur. Unfortunately, the protein complex has not been crystallized, so with only indirect evidence in hand, mechanistic conclusions need to be viewed with due caution.
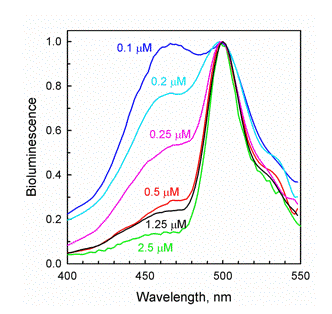
Figure 27. The bioluminescence of the
Ca2+-regulated photoprotein clytin has a single maximum at 470 nm, similar to obelin (Figure 11). The inclusion of as little as 0.1 µM of Clytia GFP in the reaction mixture reveals a green bioluminescence contribution. The spectrum is "titrated" by further additions of GFP, and by 2.5 µM the bioluminescence is almost identical to the GFP fluorescence (see Markova et al., 2010).
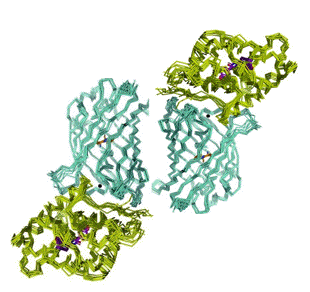
Figure 28. Transient complex of the Clytia GFP dimer (blue) associated with two clytin photoproteins (green). In each monomer the donor-acceptor separation is 45 Å (adapted from Titushin et al., 2010).
SUMMARY and PROSPECTS
The two remarkable properties of bioluminescence proteins are, their ability to convert chemical energy into light with such high efficiency, and the tuning of the color over the visible spectral range. Over the past two decades the determination of the spatial structures of several bioluminescence proteins has led to significant advances in understanding the mechanism of color tuning, contrasting with the still challenging mystery of the efficient chemical generation of products in their excited states. The bioluminescence systems of firefly, the bioluminescent bacteria, and the Ca2+-regulated photoproteins, are the ones most investigated over the years, and structural studies are shown here to provide a sound, although still qualitative, understanding of the mechanisms of bioluminescence color variation. These mechanisms are ones well-understood from solution fluorescence studies, i.e., environmental polarity, FRET, and excited state proton transfer. The non-isotropic environment of a fluorophore bound in the protein cavity in contrast to its isotropic surroundings in solution, is a barrier to a more quantitative accounting of the effect on energy levels. More developed computational methods as well as the structures of many color mutants, will greatly advance this field.
Bioluminescence proteins enjoy a multitude of applications in medical diagnostics and other areas of biomedical research, and even in entertainment. Bioluminescence methods have advantages of extreme sensitivity, do not involve radioactivity, and thus eliminate disposal requirements, and are relatively inexpensive. With the availability of all the bioluminescence color mutants illustrated here, one anticipates that multicolor imaging applications, and gene expression and clinical diagnostic kits, will be available in the near future (Ohmiya, 2014).
FRET and BIOLUMINESCENCE FRETA bit of history is helpful in understanding the mechanism of FRET (Clegg, 2006). Early in the 20th century, there was much interest in the use of the newly developing Quantum Mechanics theory to understand molecular spectra. One puzzling observation was that in a mixture of two molecular species under certain conditions, and either in vapor or condensed phases, light absorption only into one molecule could produce a fluorescence spectrum corresponding to the second molecule. This could occur even when the two were separated by many molecular dimensions, as much as 10 nm. Even in the gas phase, the likelihood of a collision within the excited state lifetime of the first molecule, was negligibly small. This property was called "sensitized fluorescence". In the 1930's, F. Perrin was responsible for a significant advance in exploring such observations. We can start with the analogy to radio transmission, which was well understood at that time. An antenna, a dipolar oscillator, could generate a signal, an electromagnetic wave that could be detected by another antenna at great distances, many wavelengths away. However, if the oscillator and antenna are separated much less than this wavelength, there is no electromagnetic wave to be formed, and instead the oscillator induces an electrical dipole effect in the receiver. This is called a "near-field" situation to be distinguished from "far-field" as in the radio transmission. Perrin published a comprehensive treatment of this sensitized fluorescence effect. The landmark contribution of T. Förster published in 1948, however, is distinguished by presenting a more complete analysis of sensitized fluorescence from molecules in solution, one that was directly testable by solution spectroscopy. Förster showed that the efficiency with which the antenna or acceptor molecule, would radiate its own fluorescence in competition with that of the donor, was a function of measurable parameters (Visser and Rolinski, 2010): the fluorescence quantum yield of the donor in the absence of acceptor Ø0, the refractive index of the solvent n, an orientation parameter 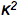 , derived from the relative orientations of the excited state transition moments of donor and acceptor, the donor-acceptor separation, and the spectral overlap of the donor fluorescence with the acceptor absorption J. The "Förster distance" R0, is that separation where the donor fluorescence and energy transfer to the acceptor are equally probable, and is obtained from the "Förster equation": , derived from the relative orientations of the excited state transition moments of donor and acceptor, the donor-acceptor separation, and the spectral overlap of the donor fluorescence with the acceptor absorption J. The "Förster distance" R0, is that separation where the donor fluorescence and energy transfer to the acceptor are equally probable, and is obtained from the "Förster equation":

Förster's 1948 paper is one of the most cited to this day and consequently, the acronym FRET is used to stand for "Förster Resonance Energy Transfer". In the literature it is sometimes referred to as "Fluorescence Resonance Energy Transfer", but this is misleading, as fluorescence is not transferred, only used to detect the energy transfer after it has occurred. There is no photon or electromagnetic wave occurring in this near-field situation. For the same reason, BRET for "Bioluminescence Resonance Energy Transfer" is incorrect, no bioluminescence is transferred from the donor, photoprotein or luciferase, to the acceptor the antenna protein. Under the appropriate conditions, the bioluminescence reaction produces excitation of the antenna protein, detected as the fluorescence of the antenna, in the known cases of lumazine protein or GFP. Therefore this phenomenon is more correctly called "Bioluminescence FRET", because the mechanism is analogous to the near-field situation in FRET. FRET is observed for donor and acceptor molecules in solution only when their concentration is in the millimolar range. At this concentration it can be easily calculated that the average distance between molecules is about 5 nm, fulfilling the distance requirement for efficient FRET, providing the other parameters are favorable. Figure 26 however, shows that the Bioluminescence FRET is seen when the bioluminescence proteins, photoprotein and GFP in this case, are at micromolar concentration, where their random separation would be greater than 100 nm, too far for FRET to occur. This means that they must be confined within a photoprotein-GFP complex. This was the origin for the supposition that such a complex had to occur and the spatial structures of the protein complexes shown in Figures 24 and 27 now support this conjecture, and significantly advance our understanding of Bioluminescence FRET mechanisms. |
THE PROTEIN DATA BANKThe X-ray crystallography method involves directing X-rays of wavelength usually 1.54 Å, through the protein crystal. The atoms of the crystal scatter the X-rays to form a diffraction pattern. Very powerful mathematical and computer methods have been developed to transform this pattern back into an electron density map of the molecule onto which the atomic positions can be accurately fitted. The protein structures shown here in this module can be viewed and manipulated interactively using free software listed at the internet site containing the Protein Data Bank. Select SEARCH, then RCSB PDB, type in the PDB code given in the legends of Figures 7, 17, or 20, then select a Display Option. The software for viewing the structures will need to be installed first, and there are detailed instructions on the web page for doing this. The structures shown in Figures 17 and 20, have been obtained using the software "Protein Explorer". (Martz, 2002. Protein Explorer : Easy Yet Powerful Macromolecular Visualization, Trends Biochem Sci 27:107-109.). |
REFERENCES
Branchini, BR. (2008). Firefly Bioluminescence, on Photobiological Sciences Online (KC Smith, ed.). American Society for Photobiology, http://www.photobiology.info/.
Branchini BR, Murtiashaw MH, Magyar RA, Portier NC, Ruggiero MC, Stroh, JG. (2002) Yellow-green and red firefly bioluminescence from 5,5-dimethyloxyluciferin. J Amer Chem Soc 124: 2112-2113.
Campbell ZT, Weichsel A, Montfort WR, Baldwin TO. (2009) Crystal structure of the bacterial luciferase/flavin complex provides insight into the function of the beta subunit. Biochemistry 48: 6085-6094.
Chung LW, Hayashi S, Lundberg M, Nakatsu T, Kato H, Morokuma K. (2008). Mechanism of efficient firefly bioluminescence via adiabatic transition state and seam of sloped conical intersection. J Am Chem Soc 130: 12880-12881.
Clegg RM. (2006) The History of FRET, in Reviews in Fluorescence, Vol. 3, eds, Geddes CD and Lakowicz JR, eds.) Springer, New York. Chap. 1, pp. 1-45.
Daubner SC, Astorga AM, Leisman GB, Baldwin TO. (1987) Yellow light emission of vibrio fischeri strain Y-1: Purification and characterization of the energy-accepting yellow fluorescent protein. Proc NatI Acad Sci USA 84: 8912-8916.
Deng L, Vysotski ES, Liu Z-J, Markova SV, Malikova NP, Lee J, Rose J, Wang B-C. (2001) Structural basis for the emission of violet bioluminescence from a W92F obelin mutant. FEBS Lett 506: 281-285.
Eremeeva EV, Markova SV, Westphal AH, Visser AJ, van Berkel WJ, Vysotski ES. (2009) The intrinsic fluorescence of apo-obelin and apo-aequorin and use of its quenching to characterize coelenterazine binding. FEBS Lett 583: 1939-1944.
Eremeeva EV, Markova SV, Frank LA, Visser AJ, van Berkel WJ, Vysotski ES. (2013) Bioluminescence and spectroscopic properties of His-Trp-Tyr triad mutants of obelin and aequorin. Photochem Photobiol Sci 12: 1016-1024.
Frank LA, Borisova VV, Markova SV, Malikova NP, Stepanyuk GA, Vysotski ES. (2008) Violet and greenish photoprotein obelin mutants for reporter applications in dual-color assay. Anal Bioanal Chem 391: 2891-2896.
Hastings JW, Schultz W, Liu L. (2009) Dinoflagellate Bioluminescence and its Circadian Regulation, on Photobiological Sciences Online (KC Smith, ed.) American Society for Photobiology, http://www.photobiology.info/.
Lee J. (2015) Bioluminescence, the Nature of the Light. (University of Georgia).
Lee J, Matheson IBC, Müller F, O'Kane DJ, Vervoort J, Visser AJWG. (1990) The mechanism of bacterial bioluminescence. In: Flavins and Flavoproteins (F Müller, ed.) CRC Press, Boca Raton, Florida Vol. II, pp. 109-151.
Liu ZJ, Vysotski ES, Deng L, Lee J, Rose J, Wang BC. (2003) Atomic resolution structure of obelin: soaking with calcium enhances electron density of the second oxygen atom substituted at the C2-position of coelenterazine. Biochem Biophys Res Commun 311: 433-439.
Malikova NP, Stepanyuk GA, Frank LA, Markova SV, Vysotski ES, Lee J. (2003) Spectral tuning of obelin bioluminescence by mutations of Trp92. FEBS Lett 554: 184-188.
Markova SV, Burakova LP, Frank LA, Golz S, Korostileva KA,Vysotski ES. (2010) Green-fluorescent protein from the bioluminescent jellyfish Clytia gregaria: cDNA cloning, expression, and characterization of novel recombinant protein, Photochem Photobiol Sci 9: 757-765.
Matz MV, Lukyanov KA, Lukyanov SA. (2002) Family of the green fluorescent protein: journey to the end of the rainbow. Bioessays 24: 953-959.
Nakatsu T, Ichiyama S, Hiratake J, Saldanha A, Kobashi N, Sakata K, Kato H. (2006) Structural basis for the spectral difference in luciferase bioluminescence. Nature 440: 372-376.
Natashin PV, Markova SV, Lee J, Vysotski ES, Liu Z-J. (2014) Crystal structures of the F88Y obelin mutant before and after bioluminescence provide molecular insight into molecular tuning among hydromedusan photoproteins. FEBS J, Jan 22.
Naumov P, Ozawa Y, Ohkubo K, Fukuzumi S. (2009) Structure and spectroscopy of oxyluciferin, the light emitter of the firefly bioluminescence. J Am Chem Soc 131: 11590-11605.
O'Kane DJ, Woodward B, Lee J, Prasher DC. (1991) Borrowed proteins in bacterial bioluminescence. Proc Natl Acad Sci USA 88: 1100-1104.
Ohmiya Y. (2014) Applications of bioluminescence. Cell based assays and imaging Photobiological Sciences Online (KC Smith, ed.) American Society for Photobiology http://www.photobiology.info/
Petushkov V, Gibson BG, Lee J. (1996) Direct measurement of excitation transfer in the protein complex of bacterial luciferase hydroxyflavin and the associated yellow fluorescence proteins from vibrio fischeri Y-1. Biochemistry 35: 8413-8418.
Sato Y, Shimizu S, Ohtaki A, Noguchi K, Miyatake H, Dohmae N, Sasaki S, Odaka M, Yohda M. (2010) Crystal structures of the lumazine protein from photobacterium kishitanii in complexes with the authentic chromophore, 6,7-dimethyl-8-(1'-D-ribityl) lumazine and its analogues, riboflavin and FMN, at high resolution. J Bacteriol 192: 127-133; 1749.
Shimomura O. (2005) The discovery of aequorin and green fluorescent protein. J Microsc 217: 3-15.
Stepanyuk GA, Golz S, Markova SV, Frank LA, Lee J, Vysotski ES. (2005) Interchange of aequorin and obelin bioluminescence color is determined by substitution of one active site residue of each photoprotein. FEBS Lett 579: 1008-1014.
Titushin MS, Feng Y, Stepanyuk GA, Li Y, Markova SV, Golz S, Wang B-C, Lee J, Wang J, Vysotski ES, Liu Z-J. (2010) NMR-derived topology of a GFP-photoprotein energy transfer complex. J Biol Chem 285: 40891-40900.
Titushin MS, Feng Y, Lee J, Vysotski ES, Liu Z-J. (2011) Protein-protein complexation in bioluminescence. Protein & Cell 2: 957-972.
van Oort B, Eremeeva EV, Koehorst RB, Laptenok SP, van Amerongen H, van Berkel WJ, Malikova NP, Markova SV, Vysotski ES, Visser AJ, Lee J. (2009) Picosecond fluorescence relaxation spectroscopy of the calcium-discharged photoproteins aequorin and obelin. Biochemistry 48: 10486-10491.
Vieira J, da Silva LP, Esteves da Silva JCG. (2012) Advances in the knowledge of light emission by firefly luciferin and oxyluciferin. J Photochem Photobiol B: Biol 117: 33-39.
Visser AJW and Rolinski OJ. (2010) Basic Photophysics, on Photobiological Sciences Online (KC Smith, ed.). American Society for Photobiology, http://www.photobiology.info/.
Viviani VR. (2009) Terrestrial Bioluminescence, on Photobiological Sciences Online (KC Smith, ed.) American Society for Photobiology, http://www.photobiology.info/.
Viviani VR, Amaral DT, Neves DR, Simoes A, Anoldi FGC. (2013) The luciferin binding site residues C/T311 (S314) influence the bioluminescence color of beetle luciferases through main chain luciferases through main-chain interaction with oxyluciferin phenolate. Biochemistry 52:19-27.
Vysotski ES and Lee J. (2004) Ca2+-regulated photoproteins: Structural insight into the bioluminescence mechanism. Acc Chem Res 37: 405-415.
Ward WW. (1998) Biochemical and physical properties of green fluorescent protein. In, Green Fluorescent Protein (M Chalfie, S Kain, eds.) Wiley-Liss, New York, pp 45-75.
Zimmer M. (2010) Green Fluorescent Protein: a Molecular Microscope, on Photobiological Sciences Online (KC Smith, ed.). American Society for Photobiology, http://www.photobiology.info/.
07/09/08
09/29/09
02/20/11
03/17/14