BASIC PHOTOCHEMISTRY
Kendric C. Smith
Emeritus Professor, Radiation Oncology (Radiation Biology)
Stanford University School of Medicine
800 Blossom Hill Road, Unit R169, Los Gatos, CA 95032
kendric@stanford.edu
web.stanford.edu/~kendric/
Photochemistry is the underlying mechanism for all of photobiology. When a molecule absorbs a photon of light, its electronic structure changes, and it reacts differently with other molecules. The energy that is absorbed from light can result in photochemical changes in the absorbing molecule, or in an adjacent molecule (e.g., photosensitization). The energy can also be given off as heat, or as lower energy light, i.e., fluorescence or phosphorescence, in order to return the molecule to its ground state. Each type of molecule has a different preference for which of these different mechanisms it uses to get rid of absorbed photon energy, e.g., some prefer fluorescence over chemistry.
The Basic Laws of Photochemistry
The First Law of Photochemistry states that light must be absorbed for photochemistry to occur. This is a simple concept, but it is the basis for performing photochemical and photobiological experiments correctly. If light of a particular wavelength is not absorbed by a system, no photochemistry will occur, and no photobiological effects will be observed, no matter how long one irradiates with that wavelength of light.
The Second Law of Photochemistry states that for each photon of light absorbed by a chemical system, only one molecule is activated for a photochemical reaction. This law is true for ordinary light intensities, however, with high-powered lasers, two-photon reactions can occur, i.e., the molecule is raised to a higher energy state than produced by single-photon absorption.
The Bunsen-Roscoe Law of Reciprocity states that a photochemical effect is directly proportional to the total energy dose, irrespective of the time required to deliver the dose. This law is true for chemicals in a test tube, but the response of cells to radiation usually involves a sequence of interacting biological reactions, making a linear "dose x time" relationship highly unlikely. There is no reciprocity when damage is produced, e.g., DNA damage, but there can be reciprocity over a narrow range of doses for photoreceptors that trigger a response, such as phytochrome (see module on Basic Photomorphogenesis).
Electromagnetic Radiation
Electromagnetic radiation consists of waves of electric and magnetic fields traveling in space at right angles to one another (Figure 1).
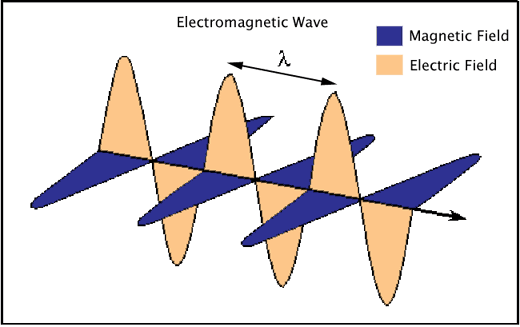
Figure 1. An electromagnetic wave showing the perpendicularly-oriented waves of electric and magnetic fields, and the characteristic wavelength (λ) of the radiation.
The electromagnetic spectrum is composed of different wavelengths of light having different photon energies, and is classified into the regions shown in Figure 2. Note that the regions of interest for photochemistry, i.e., visible and ultraviolet (UV), are only a small part of the full electromagnetic spectrum. Longer wavelengths, e.g., far infrared, tend to cause the vibrational excitation of molecules, which results in heating. Shorter wavelength X-rays cause ionization.
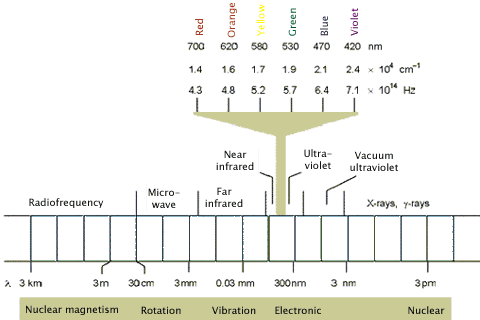
Figure 2. The electromagnetic spectrum highlighting the visible region, which along with the ultraviolet region, is capable of producing photochemical changes in molecules.
The Jablonski Diagram
The energy gained by a molecule when it absorbs a photon causes an electron to be promoted to a higher electronic energy level. Figure 3 illustrates the principal photophysical radiative and non-radiative processes displayed by organic molecules in solution. The symbols So, S1, T2, etc., refer to the ground electronic state (So), first excited singlet state (S1), second excited triplet state (T2), and so on. The horizontal lines represent the vibrational levels of each electronic state. Straight arrows indicate radiative transitions, and curly arrows indicate non-radiative transitions. The boxes detail the electronic spins in each orbital, with electrons shown as up and down arrows, to distinguish their spin.
Note that all transitions from one electronic state to another originate from the lowest vibrational level of the initial electronic state. For example, fluorescence occurs only from S1, because the higher singlet states (S2, etc.) decay so rapidly by internal conversion that fluorescence from these states cannot compete.
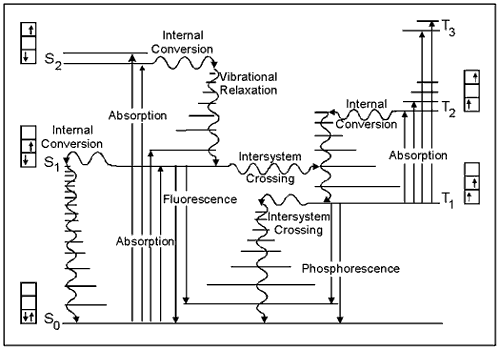
Figure 3. The basic concepts of this Jablonski diagram are presented in the Basic Photophysics module. This version emphasizes the spins of electrons in each of the singlet states (paired, i.e., opposite orientation, spins) compared to the triplet states (unpaired, i.e., same orientation, spins).
Electronically Excited States
The absorption of a UV or visible photon by a molecule produces an electronically excited state. The distribution of the electrons surrounding the nuclei change, as well as the forces between the atomic nuclei of a molecule. As a result, molecules in electronically excited states often have very different chemical and physical properties than their electronic ground states. For example,
The Beer-Lambert Law
The absorption of photons of light is described by the Beer-Lambert Law, a relationship that indicates a decrease in intensity as a beam passes through a medium that can absorb it. Consider a parallel beam of monochromatic light of initial intensity, lo, passing through a homogeneous absorbing medium (Figure 4).
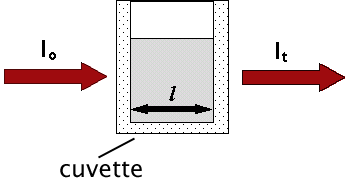
Figure 4. Schematic representation showing that light of initial intensity, lo, passing through an absorbing medium in a cuvette with light path, l, will emerge with a final intensity, lt.
In spectroscopy, absorbance, A, and Optical Density, OD, are used somewhat interchangeably. Optical Density can be expressed as:
where T is the transmittance. It can also be expressed as:
Note the log scale. An OD of 1 will have a transmittance of 0.1, and a
% transmission of 10, but at OD = 2, the transmittance is 0.01, and the % transmission = 1.
Another way of expressing this information is to use the Beer-Lambert Law. It states that the absorbance, A, of a molecular species is linearly related to the path length (centimeter), l, the absorber concentration (moles/liter), c, and the proportionality constant,
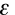 , called the molar extinction coefficient of the absorbing molecular species (liters/mole-cm) [a measure of how strongly a chemical species absorbs light at a given wavelength].
cl
, called the molar extinction coefficient of the absorbing molecular species (liters/mole-cm) [a measure of how strongly a chemical species absorbs light at a given wavelength].
cl
Energy Level Diagram
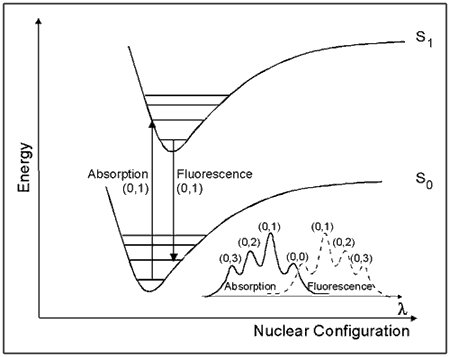
One way to view the properties of molecular excited states is shown by the potential energy diagram in Figure 5. This diagram, known as a Franck-Condon energy level diagram, shows potential energy curves for the ground state (So), and first excited singlet state (S1) of an organic molecule as a function of nuclear configuration. These curves are sometimes referred to as potential energy wells, because of their shape. The horizontal lines within each curve represent the vibrational levels of each electronic state. The lowest vibrational state for each energy level is designated as 0, and the levels above it are successively 1, 2, etc. The band assignments in brackets (e.g., (0, 1)) indicate, respectively, the vibrational level of the initial state, and of the final state involved in a transition.
See text for details.
The horizontal axis is the nuclear configuration, which can be thought of as the distance between nuclei. When considering two atoms bonded to each other, the bottom of the well corresponds to the equilibrium bond length. Because excitation involves the movement of charge density into an antibonding orbital, the equilibrium bond length in S1 is generally longer than in S0. This is illustrated in Figure 5 by the displacement of the S1 well to the right of the S0 well.
The absorption of light takes place on a much faster time scale
(~ 10-15 s) than molecular vibration (~ 10-12 s), hence the initially formed excited state must have the same nuclear configuration as the ground state. This transition is called the vertical or Franck-Condon transition, and results in the molecule having excess vibrational energy. The excess vibrational energy can be dissipated through the process of vibrational relaxation, i.e., the process of internal conversion, which returns the molecule to the lowest vibrational level of S1. Fluorescence usually occurs from the lowest vibrational level of S1. Because these transitions occur at lower energies than absorption, fluorescence is observed at longer wavelengths (λ) than absorption (i.e., lower energy), as shown in the lower right corner of Figure 5.
Quantum Yields and Lifetimes
The energy that a molecule gains when it absorbs light is subsequently lost by a molecule in one of several ways. As shown graphically in the Jablonski diagram (Figure 3), it can lose the energy as heat as it returns to the ground state (internal conversion). Alternately, it can lose the energy as light (fluorescence), usually on a nanosecond time scale. A third pathway is intersystem crossing to a triplet state, from which energy can also be lost as light (phosphorescence), but over much longer times (microseconds or longer). And finally, the energy can be transferred to another molecule.
The quantum yield of a process is the probability that an absorbed photon undergoes one particular process. Thus, one can define a quantum yield for fluorescence, a quantum yield for phosphorescence, or a quantum yield for other pathways. Each quantum yield is typically a number between 0 and 1 (except under unusual circumstances), and the total of all quantum yields for a particular absorption event should sum to one. Note that these processes are competing. If conditions are altered such that the quantum yield for fluorescence is increased, then the quantum yield for some other process(es) must decrease.
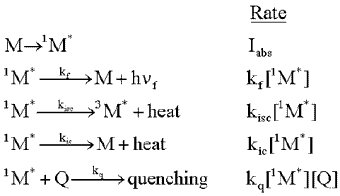
Consider a molecule, M, that is exposed to light, and absorbs photons at the rate, Iabs. As shown in the following formulas, the excited singlet state of the molecule, 1M*, can fluoresce emitting a photon, h
 f. It can lose energy as heat,
and move to the triplet excited state, 3M*, by intersystem
crossing (isc). Finally, it can lose energy as heat, and move to the ground state
by internal conversion (ic). The rate of each of these loss processes will be proportional
to the concentration of the excited singlet state, 1M*,
and a rate constant, k, for each process, as given in the second column below.
f. It can lose energy as heat,
and move to the triplet excited state, 3M*, by intersystem
crossing (isc). Finally, it can lose energy as heat, and move to the ground state
by internal conversion (ic). The rate of each of these loss processes will be proportional
to the concentration of the excited singlet state, 1M*,
and a rate constant, k, for each process, as given in the second column below.
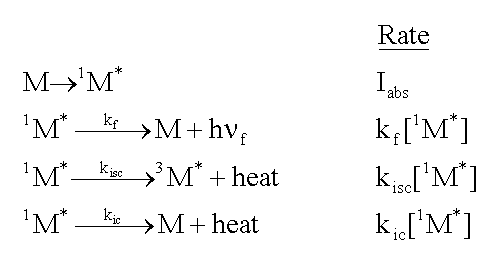
![]()
Intermolecular Processes: Excited State Quenching
When a second molecule (a quencher, Q) interacts with a molecule in an excited state, new ways may be created for the excited state species to lose its energy of excitation. Such interactions (collisions) can induce the loss of energy in the form of heat, which is called physical quenching, or it can cause the energy to be transferred to the second molecule with or without the transfer of an electron. The former is called energy transfer, and the latter, electron transfer. Formally, one can write:

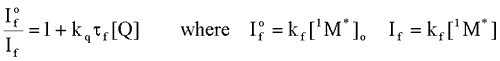
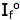 is the intensity, or rate of fluorescence, without a quencher;
is the intensity, or rate of fluorescence, without a quencher; 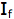 is the intensity, or rate of fluorescence, with a quencher;
is the intensity, or rate of fluorescence, with a quencher; 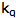 is the quencher rate coefficient;
is the quencher rate coefficient; 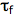 is the fluorescence lifetime of A without a quencher present, and
is the fluorescence lifetime of A without a quencher present, and 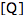 is the concentration of the quencher. This relationship is referred to as the Stern-Volmer equation.
is the concentration of the quencher. This relationship is referred to as the Stern-Volmer equation. Types of Photochemical Reactions
1. Linear addition to an unsaturated molecule, e.g., the pyrimidine base, thymne, in DNA can combine with the amino acid residue, cysteine, in proteins. This is a model for the photochemical crosslinking of DNA and proteins by UV radiation (see the Module on DNA-Protein Crosslinks).
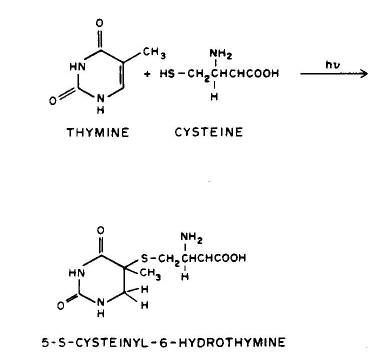
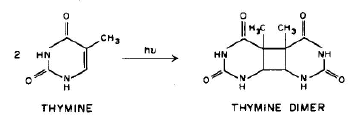
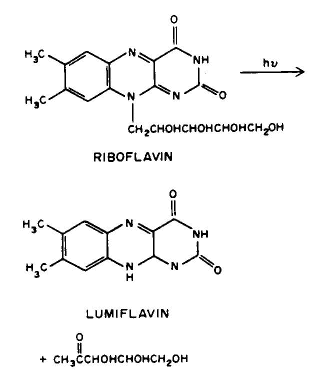
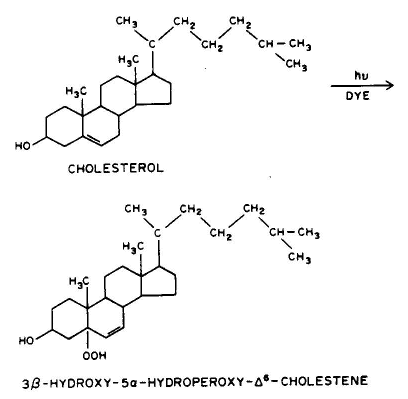
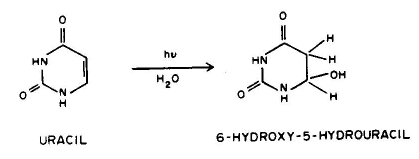
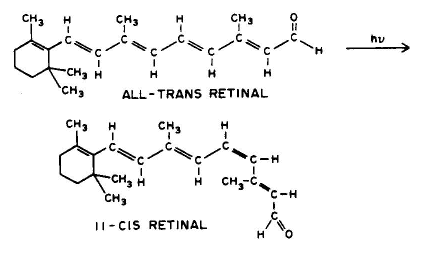
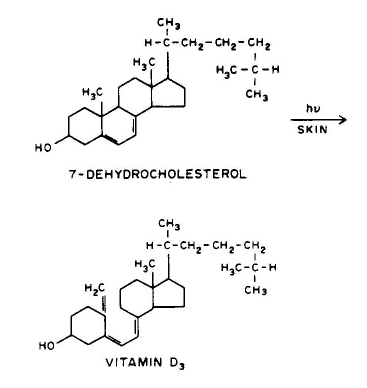
Conclusion
Additional information can be found in the Suggested Reading section, and in the Photochemistry module.
Suggested Reading
Coxon, JM and Halton, B, Organic Photochemistry, Cambridge University Press, 1987
Gilbert, A and Baggott, J, Essentials of Molecular Photochemistry, Blackwell Science Ltd, 1990.
Kagan, J, Organic Photochemistry: Principles and Applications, Academic Press, 1993.
Klessinger, M and Michl, J, Excited States and Photo-Chemistry of Organic Molecules, Wiley-VCH, 1995.
Smith, K.C., The Science of Photobiology, 2nd Ed. Plenum Pub Corp, 1989.
Turro, NT, Modern Molecular Photochemistry, University Science Books, 1991.
Wayne, CE and Wayne, RP, Photochemistry, Oxford University Press, 1996.
Online Articles
Franck-Condon Principle, Wikipedia
Virtual Textbook of Organic Chemistry, William Reusch [Note especially: "Other Topics: Photochemistry"]
UNC-Chapel Hill Chemistry Fundamentals, An Interactive Educational Exercise
Chemistry, Wikipedia
10/03/09
06/02/11
03/28/13
03/18/14